Chapitre 6 : PCR (Polymerase Chain Reaction)
La PCR est une technique très simple: tout ce qui se passe est qu’une petite région d’une molécule d’ADN, par exemple un seul gène, est copiée à plusieurs reprises par une enzyme ADN polymérase. Cela peut sembler un exercice plutôt trivial, mais il a une multitude d’applications dans la recherche génétique et dans des domaines plus larges de la biologie. Nous commencerons ce chapitre par un aperçu de la réaction en chaîne de la polymérase pour comprendre exactement ce qu’il réalise. Ensuite, nous examinerons les questions clés qui déterminent si une expérience de PCR individuelle est réussie, avant d’examiner quelques-unes des méthodes qui ont été conçues pour étudier les fragments d’ADN amplifiés qui sont obtenus.
La PCR (polymerase chaine reaction ) en détaille :
La réaction en chaîne par polymerase conduit à l’amplification sélective d’une région choisie d’une molécule d’ADN. Toute région de n’importe quelle molécule d’ADN peut être choisie, pour autant que les séquences aux frontières de la région soient connues. Les séquences de la frontière doivent être connues car, pour effectuer une PCR, deux oligonucleotides courts doivent s’hybrider à la molécule d’ADN, un à chaque brin de la double hélice . Ces oligonucleotides, qui servent d’amorces pour les réactions de synthèse d’ADN, délimitent la région qui sera amplifié. L’amplification est habituellement effectuée par l’enzyme ADN polymérase I de Thermus Aquatique. Cet organisme vit dans les sources chaudes, et bon nombre de ses enzymes, y compris la Taq polymerase, sont thermostables, ce qui signifie qu’ils sont résistants à la dénaturation par traitement thermique. Comme on le verra dans un instant, la thermostabilité de la polymérase Taq est une exigence essentielle dans la méthodologie PCR.
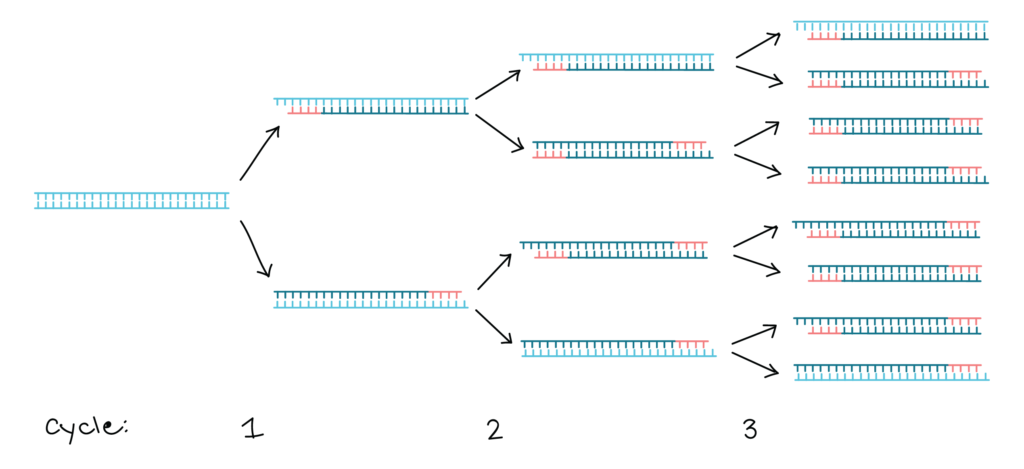
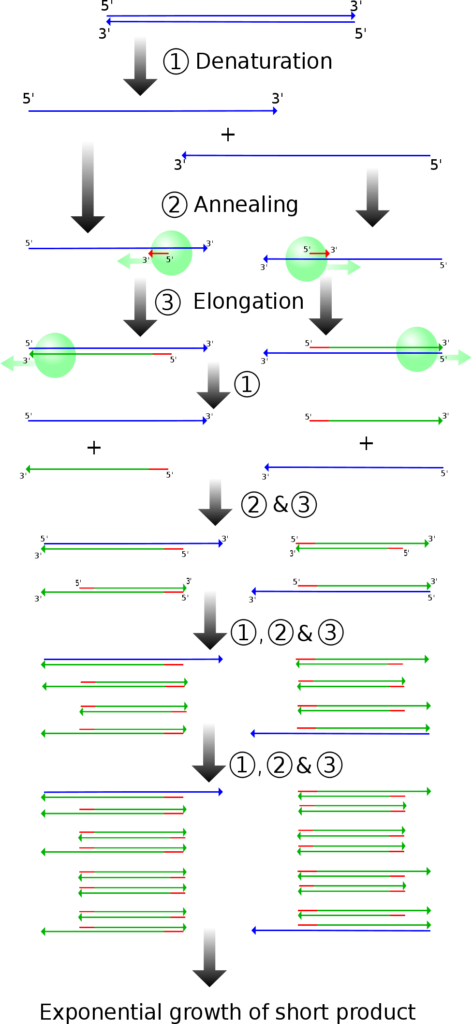
Pour réaliser une expérience de PCR, l’ADN cible est mélangé à la polymerase Taq, aux deux amorces oligonucléotidiques et à une réserve de nucleotides. La quantité d’ADN cible peut être très faible, car la PCR est extrêmement sensible et est capable de fonctionner avec une seule molécule de départ. On commence la réaction en chauffant le mélange à 94 ° C. A cette température, les liaisons hydrogène qui maintiennent ensemble les deux polynucleotides de la double hélice sont rompues, de sorte que l’ADN cible devient dénaturé en molécules monocaténaires. La température est ensuite réduite à 50-60 ° C, ce qui se traduit par une certaine réintégration des brins simples de l’ADN cible, mais permet également aux amorces de se fixer à leurs positions de recuit. La synthèse d’ADN peut maintenant commencer, de sorte que la température est élevée à 74 ° C, juste en dessous de l’optimum pour la polymerase Taq. Dans cette première étape de la PCR, un ensemble de « produits longs » est synthétisé à partir de chaque brin de l’ADN cible. Ces polynucleotides ont des extrémités 5 ‘identiques mais des extrémités 3’ aléatoires, ces dernières représentant des positions où la synthèse d’ADN se termine par hasard. Le cycle de dénaturation-recuit-synthèse est maintenant répété. Les produits longs dénaturés et les quatre brins résultants sont copiés au cours de la phase de synthèse d’ADN. Cela donne quatre molécules à double brin, dont deux sont identiques aux produits du premier cycle et dont deux sont entièrement faits d’ADN nouveau. Au cours du troisième cycle, ces derniers donnent lieu à des «produits courts» dont les extrémités 5 ‘et 3’ sont toutes deux fixées par les positions de recuit d’amorce. Dans les cycles suivants, le nombre de produits courts s’accumule de manière exponentielle (doublant à chaque cycle) jusqu’à ce que l’une des composantes de la réaction s’épuise. Cela signifie qu’après 30 cycles, il y aura plus de 130 millions de produits courts dérivés de chaque molécule de départ. En termes réels, cela équivaut à plusieurs microgrammes de produit de PCR à partir de quelques nanogrammes ou moins d’ADN cible.
A la fin d’une PCR, un échantillon du mélange réactionnel est habituellement analysé par l’électrophorèse sur gel de l’agarose , on verra qu’on a produit suffisamment d’ADN pour le fragment amplifié pour il soit visible sous la forme d’une bande discrète après coloration au bromure d’éthidium. Cela peut en soi fournir des informations utiles sur la région de l’ADN qui a été amplifiée ou, le produit de PCR peut être examiné par des techniques telles que le séquençage d’ADN.
Concevoir les amorces oligonucléotidiques pour une PCR:
Les amorces sont la clé du succès ou de l’échec d’une expérience PCR. Si les amorces sont conçues correctement, l’expérience conduit à l’amplification d’un seul fragment d’ADN, correspondant à la région cible de la molécule modèle. Si les amorces sont mal conçues, l’expérience échouera, peut-être parce qu’aucune amplification ne se produit, ou peut-être parce que le mauvais fragment ou plus d’un fragment est amplifié. De toute évidence, beaucoup de réflexion doit être placée dans la conception des amorces. L’élaboration de séquences appropriées pour les amorces n’est pas un problème: elles doivent correspondre aux séquences flanquant la région cible sur la molécule modèle.

Chaque amorce doit naturellement être complémentaire (non identique) à son brin matrice pour que l’hybridation se produise, et les extrémités 3 ‘des amorces hybridées doivent pointer l’une vers l’autre. Le fragment d’ADN à amplifier ne doit pas être supérieur à environ 3 kb de longueur et idéalement moins de 1 kb. Des fragments allant jusqu’à 10 kb peuvent être amplifiés par des techniques de PCR standard, mais plus le fragment est long, moins l’amplification est efficace et plus il est difficile d’obtenir des résultats cohérents. L’amplification de fragments très longs – jusqu’à 40 kb est possible, mais nécessite des méthodes spéciales. La première question importante à aborder est la longueur des amorces. Si les amorces sont trop courtes, elles pourraient s’hybrider à des sites non cibles et donner des produits d’amplification indésirables. Pour illustrer ce point, imaginez que l’ADN humain total est utilisé dans une expérience de PCR avec une paire d’amorces de huit nucléotides de longueur (dans le jargon de la PCR, on parle de «8-mères»). Le résultat probable est qu’un nombre de fragments différents seront amplifiés. En effet, on s’attend à ce que les sites de fixation de ces amorces se produisent, en moyenne, une fois tous les 48 = 65 536 pb, ce qui donne approximativement 49 000 sites possibles dans la séquence nucléotidique de 3 200 000 kb qui constitue le génome humain. Cela signifie qu’il serait très peu probable qu’une paire d’amorces 8-mères donnerait un seul produit d’amplification spécifique avec de l’ADN humain.
Que se passe-t-il si on utilise des amorces 17-mères ? La fréquence attendue d’une séquence 17-mères est une fois tous les 417 = 17 179 869 184 pb. Ce chiffre est plus de cinq fois supérieur à la longueur du génome humain, si bien qu’un amorce 17-mère devrait avoir un seul site d’hybridation dans l’ADN humain total. Une paire d’amorces 17-mer devrait donc donner un seul produit d’amplification spécifique . Pourquoi ne pas simplement faire les amorces aussi longtemps que possible? La longueur de l’amorce influe sur la vitesse à laquelle elle s’hybride à l’ADN matrice, les amorces longues s’hybridant à une vitesse plus lente. L’efficacité de la PCR, mesurée par le nombre de molécules amplifiées produites au cours de l’expérience, est donc réduite si les amorces sont trop longues, car une hybridation complète aux molécules matrices ne peut se produire dans le temps imparti pendant le cycle réactionnel. En pratique, les amorces plus longues que 30-mers sont rarement utilisées.
Working out the correct temperatures to use :
Au cours de chaque cycle de PCR, le mélange réactionnel est transféré entre trois températures:
1) La température de dénaturation, habituellement 94 ° C, qui casse les paires de bases et libère l’ADN monocaténaire pour servir de modèle pour la synthèse de nouvel ADN.
2) La température d’hybridation ou de recuit, à laquelle les amorces se fixent l’ADN:
La température de recuit (hybridation) est la plus importante car, encore une fois, cela peut affecter la spécificité de la réaction. L’hybridation ADN-ADN est un phénomène. Si la température est trop élevée, aucune hybridation n’a lieu et au lieu de cela les amorces et les brins modèles restent dissociés. Toutefois, si la température est trop basse et peu correspondante, dans lesquels toutes les paires de bases formés sont stables et aussi les mismatches. Si cela se produit, les calculs antérieurs concernant les longueurs appropriées pour les amorces deviennent non pertinentes, car ces calculs supposaient que seuls les hybrides parfaits d’amorce-modèle peuvent se former. Si des mismatches (inadéquations) sont tolérées, Le nombre de sites d’hybridation potentiels pour chaque amorce est grandement augmenté, et l’amplification est plus susceptible de se produire à des sites non cibles dans la molécule modèle.La température de recuit idéale doit être suffisamment faible pour permettre l’hybridation entre l’amorce et le model, mais suffisamment élevée pour empêcher les mismatches de former l’ADN. Cette température peut être estimée en déterminant la melting température (fusion) et ctte température ou la Tm de l’hybride amorce-matrice. La Tm est la température à laquelle l’hybride correctement formé se dissocie. Une température de 1-2 °C en dessous de cette température être suffisamment faible pour permettre la formation de l’hybride primaire-modèle correctement, mais elle est trop elevée pour qu’ un hybride avec une seule inadéquation puisse pas être stable. La Tm peut être déterminé expérimentalement mais elle est souvent calculée à partir de la formule simple : Tm = (4 × [G + C]) + (2 × [A + T])°C dans laquelle [G + C] est le nombre de nucléotides G et C dans la séquence d’amorce, et [A + T] est le nombre de nucléotides A et T. La température d’hybridation (recuit) pour une expérience de PCR est donc déterminée en calculant La Tm pour chaque amorce et en utilisant une température de 1-2 ° C en dessous de ce chiffre. Remarquez que cela signifie que les deux amorces doivent être conçues de telle sorte qu’elles aient des Tms identiques. Si ce n’est pas le cas, la température d’hybridation( recuit) appropriée pour une amorce peut être trop élevée ou trop faible pour l’autre amorce.
3) La température d’extension, à laquelle se produit la synthèse d’ADN. Il est généralement fixé à 74 ° C, juste en dessous de l’optimum pour la polymerase Taq.
Les produits de PCR:
La PCR est souvent le point de départ d’une série d’expériences dans lesquelles le produit d’amplification est étudié de diverses manières afin d’obtenir des informations sur la molécule d’ADN qui a servi de modèle d’origine. Bien qu’un large éventail de procédures a été conçu pour étudier les produits de PCR, trois techniques sont particulièrement importantes:
l) l’ électrophorèse sur gel des produits de PCR
2) Le clonage des produits de PCR
3) Le séquençage des produits de PCR.
L’ électrophorèse sur gel des produits de PCR :
Les résultats de la plupart des expériences de PCR sont vérifiés en faisant tourner une partie du mélange réactionnel amplifié dans un gel d’agarose. Une bande représentant l’ADN amplifié peut être visible après coloration, ou si le rendement en ADN est faible, le produit peut être détecté par la méthode d’hybridation de Southern. Si la bande attendue est absente, ou si des bandes supplémentaires sont présentes, quelque chose a mal tourné et l’expérience doit être répétée. Dans certains cas, l’électrophorèse sur gel d’agarose est utilisée non seulement pour déterminer si une expérience de PCR a fonctionné, mais aussi pour obtenir des informations supplémentaires. Par exemple, la présence de sites de restriction dans la région amplifiée de l’ADN matrice peut être déterminée en traitant le produit de PCR avec une endonucléase de restriction avant de faire tourner l’échantillon dans le gel d’agarose.
Il s’agit d’un type d’analyse du polymorphisme de la longueur des fragments de restriction (RFLP) et est important à la fois dans la construction des cartes du génome et dans l’étude des maladies génétiques . D’autre part, la taille exacte du produit de PCR peut être utilisée pour établir si l’ADN matrice contient une mutation d’insertion ou de déletion dans la région amplifiée . Les mutations de longueur de ce type forment la base du profilage d’ADN, une technique centrale en médecine légale. Dans certaines expériences, la simple présence ou absence du produit de PCR est la caractéristique diagnostique. Un exemple est lorsque la PCR est utilisée comme procédé de criblage pour identifier un gène désiré à partir d’une banque génomique ou d’ADNc. La réalisation de PCR avec chaque clone dans une bibliothèque génomique peut sembler une tâche fastidieuse, mais un des avantages de la PCR est que les expériences individuelles sont rapides à mettre en place et de nombreuses PCR peuvent être effectuées en parallèle. La charge de travail peut également être réduite par un criblage combinatoire, dont un exemple est illustré à la figure.
Clonage des produits de PCR :
Certaines applications nécessitent qu’après une PCR, les produits résultants soient ligaturés à un vecteur et examiné par l’une quelconque des méthodes standard utilisées pour étudier l’ADN cloné. Cela peut sembler facile, mais il y a des complications.
Le premier problème concerne les extrémités des produits de PCR. D’un examen de Figure ci dessous, on pourrait imaginer que les produits courts résultant de l’amplification par PCR Sont à extrémités franches. Si tel était le cas, ils pourraient être insérés dans un vecteur de clonage par une ligature à extrémités franches ou, en variante, les produits de PCR pourraient être munis d’extrémités collantes Par la fixation d’éléments de liaison ou d’adaptateurs .
Malheureusement, la situation n’est pas simple. La Taq polymerase tend à ajouter un nucleotide supplémentaire, habituellement une Adénosine, à la fin de chaque brin qu’il synthétise. Cela signifie qu’un produit double-brin de PCR n’est pas à extrémités franches, et la plupart des extrémités 3 ‘ ont une extrémité unique.

Les surplombs pourraient être enlevés par traitement avec une enzyme exonucléase, ce qui donne des produits de PCR avec de vraies extrémités franches, mais ce n’est pas Une approche populaire car il est difficile d’empêcher l’exonucléase de devenir hyperactive et provoquer des dommages supplémentaires aux extrémités des molécules.
Une solution consiste à utiliser un vecteur de clonage spécial qui porte des surplombs de thymidine (T) et qui peut donc être ligaturé à un produit de PCR .

Ces vecteurs sont Habituellement préparé en restreignant un vecteur standard au niveau d’un site à extrémités franches, puis en traitant Avec Taq polymerase en présence de 2′-désoxythymidine 5′-triphosphate (dTTP). Aucune amorce n’est présente ainsi la seule chose que la polymérase puisse faire est d’ajouter un nucléotide T aux extrémités 3 ‘ de la molécule de vecteur à extrémités franches, ce qui a pour résultat le vecteur à queue T dans lequel la PCR Produits peuvent être insérés. Des vecteurs spéciaux de ce type ont également été conçus pour être utilisés avec la méthode de ligation de la topoisomérase, et c’est actuellement La méthode la plus populaire de clonage des produits PCR.
Une seconde solution consiste à concevoir des amorces qui contiennent des sites de restriction. Après la PCR, les produits sont traités par l’endonucléase de restriction, qui coupe chaque molécule à l’intérieur de la séquence d’amorce, laissant des fragments à extrémité collante qui peuvent être ligaturés efficacement dans un vecteur de clonage standard. L’approche ne se limite pas aux cas où les amorces s’étendent sur des sites de restriction qui sont présents dans l’ADN matrice. Au lieu de cela, le site de restriction peut être inclus dans une courte extension à l’extrémité 5′ de chaque amorce. Ces extensions ne peuvent pas s’hybrider à la molécule modèle, Mais ils sont copiés au cours de la PCR, résultant en produits de PCR qui portent des terminaux avec des sites de restrictio
Problèmes avec le taux d’erreur de Taq polymérase :
Toutes les ADN polymérases font des erreurs pendant la synthèse de l’ADN, en introduisant des nucléotides incorrects dans le brin d’ADN croissant. La plupart des polymérases, cependant, sont capable de corriger ces erreurs en inversant l’erreur et en re-synthétisant les séquence. Cette propriété est appelée fonction « relecture » et dépend de la polymérase possédant une activité exonucléase 3′ à 5′. La Taq polymérase manque d’une activité de correction d’épreuves et, par conséquent, est incapable de corriger ses erreurs. Cela signifie que l’ADN synthétisé par la Taq polymérase n’est pas toujours une copie exacte de la molécule modèle. Le taux d’erreur a été estimé à une erreur pour chaque 9000 nucléotides d’ADN qui est synthétisé, ce qui pourrait sembler être presque Insignifiant mais qui se traduit par une erreur dans chaque 300 pb pour les produits PCR obtenu après 30 cycles. C’est parce que la PCR implique des copies de copies, de sorte que les erreurs induites par la polymérase s’accumulent progressivement, les fragments produits à la fin d’une PCR contenant des copies d’erreurs antérieures ainsi que de nouvelles erreurs présenté lors de la dernière ronde de synthèse. Pour de nombreuses applications, ce taux d’erreur élevé ne pose pas de problème. En particulier, le séquençage d’un produit de PCR fournit la séquence correcte du modèle, même si les produits de PCR contiennent les erreurs introduites par la Taq polymérase. C’est parce que les erreurs sont réparties aléatoirement, donc pour chaque molécule qui a une erreur à un nucléotide, il y aura de nombreuses molécules avec la séquence correcte. Dans ce contexte le taux d’erreur est en effet insignifiant. Ce n’est pas le cas si les produits PCR sont clonés. Chaque clone résultant contient plusieurs copies d’un seul fragment amplifié, de sorte que l’ADN cloné n’a pas nécessairement la même séquence que la molécule modèle d’origine utilisée dans la PCR. Cette possibilité confère une incertitude à toutes les expériences réalisées avec les produits clonées de PCR et dicte que, chaque fois que c’est possible, l’ADN amplifié doit être étudié directement au lieu d’être cloné.
La PCR en temps réel permet de quantifier la quantité de matière de départ :
The amount of product that is synthesized during a set number of cycles of a PCR
depends on the number of DNA molecules that are present in the starting mixture
(Table 9.1). If there are only a few DNA molecules at the beginning of the PCR then
relatively little product will be made, but if there are many starting molecules then the
product yield will be higher. This relationship enables PCR to be used to quantify the
number of DNA molecules present in an extract.
Table 9.1
Number of short products synthesized after 25 cycles of PCR with different numbers of starting molecule.
NUMBER OF STARTING MOLECULES NUMBER OF SHORT PRODUCTS
1 4,194,304
2 8,388,608
5 20,971,520
10 41,943,040
25 104,857,600
50 209,715,200
100 419,430,400
Test 1 2 3 4
9.4.1 Carrying out a quantitative PCR experiment
In quantitative PCR (qPCR) the amount of product synthesized during a test PCR is
compared with the amounts synthesized during PCRs with known quantities of starting
DNA. In the early procedures, agarose gel electrophoresis was used to make these
comparisons. After staining the gel, the band intensities were examined to identify the
control PCR whose product was most similar to that of the test (Figure 9.17). Although
easy to perform, this type of qPCR is imprecise, because large differences in the amount
of starting DNA give relatively small differences in the band intensities of the resulting
PCR products.
Today, quantification is carried out by real-time PCR, a modification of the standard
PCR technique in which synthesis of the product is measured over time, as the PCR
proceeds through its series of cycles. There are two ways of following product synthesis
in real time:
l A dye that gives a fluorescent signal when it binds to double-stranded DNA
can be included in the PCR mixture. This method measures the total amount of
double-stranded DNA in the PCR at any particular time, which may over-estimate
the actual amount of the product because sometimes the primers anneal to one
another in various non-specific ways, increasing the amount of double-stranded
DNA that is present.
l A short oligonucleotide called a reporter probe, which gives a fluorescent signal
when it hybridizes to the PCR product, can be used. Because the probe only
hybridizes to the PCR product, this method is less prone to inaccuracies caused by
primer-primer annealing. Each probe molecule has pair of labels. A fluorescent dye
Chapter 10 The Polymerase Chain Reaction 159
Figure 9.17
Using agarose gel electrophoresis to quantify the amount
of DNA in a test PCR. Lanes 1 to 4 are control PCRs
carried out with decreasing amounts of template DNA.
The intensity of staining for the test band suggests that
this PCR contained approximately the same amount of
DNA as the control run in lane 2.
Note: The numbers assume that amplification is 100% efficient, none of the reactants becoming limiting during the course of the PCR.
Quenching
compound
Fluorescent
label
Fluorescent
signal
Probe
Oligonucleotide
probe
DNA target
DNA
Figure 9.18
Hybridization of a reporter probe to its target DNA.
Amount of PCR product
Number of cycles
Threshold
Figure 9.19
Quantification by real-time PCR. The graph shows
product synthesis during three PCRs, each with a
different amount of starting DNA. During a PCR, product
accumulates exponentially, the amount present at any
particular cycle proportional to the amount of starting
DNA. The blue curve is therefore the PCR with the
greatest amount of starting DNA, and the green curve is
the one with the least starting DNA. If the amounts of
starting DNA in these three PCRs are known, then the
amount in a test PCR can be quantified by comparison
with these controls. In practice, the comparison is made
by identifying the cycle at which product synthesis
moves above a threshold amount, indicated by the
horizontal line on the graph.
is attached to one end of the oligonucleotide, and a quenching compound, which
inhibits the fluorescent signal, is attached to the other end (Figure 9.18). Normally
there is no fluorescence because the oligonucleotide is designed in such a way that
its two ends base pair to one another, placing the quencher next to the dye.
Hybridization between the oligonucleotide and the PCR product disrupts this base
pairing, moving the quencher away from the dye and enabling the fluorescent
signal to be generated.
Both systems enable synthesis of the PCR product to be followed by measuring the
fluorescent signal. Quantification again requires comparison between test and control
PCRs, usually by identifying the stage in the PCR at which the amount of fluorescent
signal reaches a pre-set threshold (Figure 9.19). The more rapidly the threshold is
reached, the greater the amount of DNA in the starting mixture.
9.4.2 Real-time PCR can also quantify RNA
Real-time PCR is often used to quantify the amount of DNA in an extract, for example
to follow the progression of a viral infection by measuring the amount of pathogen DNA
that is present in a tissue. More frequently, however, the method is used as a means of
measuring RNA amounts, in particular to determine the extent of expression of a
particular gene by quantifying its mRNA. The gene under study might be one that is
switched on in cancerous cells, in which case quantifying its mRNA will enable the
160 Part I The Basic Principles of Gene Cloning and DNA Analysis
Standard PCR
Chapitre 5 :Vecteurs de clonage pour les plantes
Ces vectors de clonage ont été développés dans les années 80 et leur utilisation a conduit à la culture des OGM (organismes génétiquement modifiées). Trois types de vecteurs ont été utilisés avec plus ou moins de succès pour le clonage des Plantes :
- Vecteurs à base de plasmides naturels d’Agrobacterium.
- Transfert direct de gène en utilisant différents types d’ADN plasmidique.
-
Vecteurs à base de virus végétaux.
Agrobacterium tumefaciens – le plus petit ingénieur génétique de la nature :
Bien qu’aucun plasmide naturel ne soit connu dans les plantes supérieures, un plasmide bactérien, le plasmide Ti d’Agrobacterium tumefaciens, est d’une grande importance. A. tumefaciens est un micro organisme du sol qui cause la maladie de la galle du collet chez de nombreuses espèces de plantes dicotylédones. La gale de la couronne se produit lorsqu’une plaie sur la tige permet à la bactérie A.Tumefaciens d’envahir la plante. Après l’infection, les bactéries provoquent une prolifération du tissu de la tige dans la région de la couronne. La capacité de causer la maladie de la galle de la couronne est associée à la présence du plasmide Ti (tumeur Induisant) dans la cellule bactérienne. Il s’agit d’un grand plasmide (plus de 200 kb) qui porte de nombreux gènes impliqués dans le processus infectieux. Une caractéristique remarquable du plasmide Ti est que, après infection, une partie de la molécule est intégrée dans l’ADN chromosomique de la plante . Ce segment, appelé T-ADN, est entre 15 et 30 kb, selon la souche. Il est maintenu sous une forme stable dans la cellule végétale et est transmise à des cellules filles en tant que partie intégrante des chromosomes.
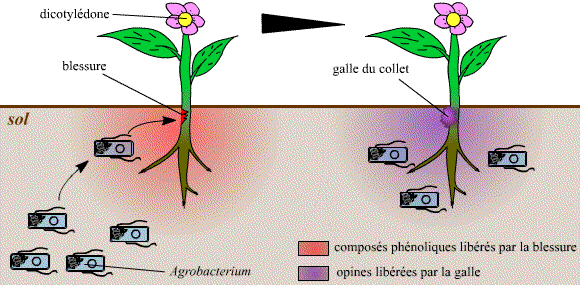
Mais la caractéristique la plus remarquable du plasmide Ti est que l’ADN-T contient Huit gènes qui sont exprimés dans la cellule végétale et sont responsables des Propriétés cancereuses des cellules transformées. Ces gènes dirigent également la synthèse de Composés, appelés opines, que les bactéries utilisent comme nutriments. En bref, A. tumefaciens modifie génétiquement la cellule végétale pour ses propres fins.
Utilisation du plasmide Ti pour introduire de nouveaux gènes dans une cellule végétale :
Il a été réalisé très rapidement que le plasmide Ti pouvait être utilisé pour transporter de nouveaux gènes dans des cellules végétales. Tout ce qui serait nécessaire serait d’insérer les nouveaux gènes dans l’ADN-T et ensuite la bactérie pourrait faire le travail acharné pour les intégrer dans l’ADN chromosomique de la plante. En pratique, cela s’est avéré une proposition délicate, principalement parce que la grande taille du plasmide Ti rend la manipulation de la molécule très difficile. Le problème principal est, bien entendu, qu’un site de restriction unique est impossible avec un plasmide de 200 kb. De nouvelles stratégies doivent être développées pour l’insertion d’un nouvel ADN dans le plasmide. Les deux techniques suivantes sont actuellement utilisées :
La stratégie du vecteur binaire :
elle est basée sur l’observation que le L’ADN-T n’a pas besoin d’être physiquement fixé au reste du plasmide Ti. Un système à deux plasmides, avec l’ADN-T sur une molécule relativement petite, et le reste sur un plasmide sous sa forme normale, est tout aussi efficace à la transformation des cellules végétales. En fait, certaines souches de A. tumefaciens et les autres agro-bactéries de la famille ont normalement des systèmes plasmidiques binaires. Le plasmide T-ADN est suffisamment petit pour avoir un seul site de restriction et d’être manipulé en utilisant des techniques standards.
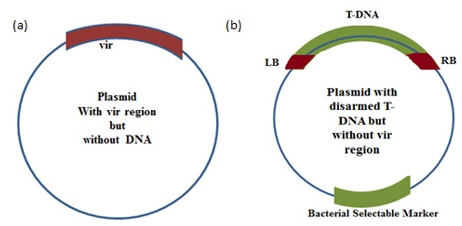
La stratégie de co-intégration:
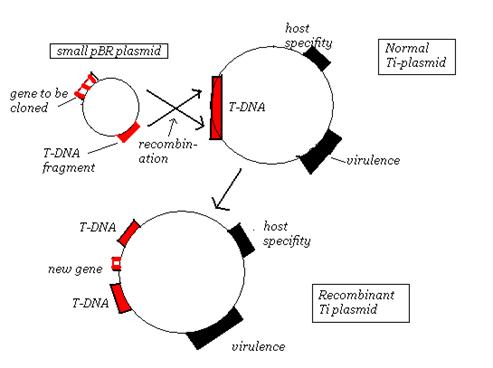
celle-ci utilise un plasmide entièrement nouveau, basée sur un vecteur d’E. coli, mais portant une petite partie de l’ADN-T. L’homologie entre la nouvelle molécule et le plasmide Ti signifie que si les deux sont présents dans la même cellule d’ A. tumefaciens, la recombinaison peut intégrer le plasmide E. coli dans La région T-ADN. Le gène à cloner est donc inséré dans un site de restriction sur le petit plasmide E. coli, introduit dans des cellules de A. tumefaciens Portant un plasmide Ti, et le processus de recombinaison naturelle fait le nécessaire pour intégrer le nouveau gène dans l’ADN-T. L’infection de la plante conduit à l’insertion du nouveau gène, avec le reste de l’ADN-T, dans les chromosomes végétaux.
Production de plantes transformées avec le plasmide Ti:
Si des A. tumefaciens contenant un plasmide Ti modifié sont introduites dans une plante d’une manière naturelle, par l’infection d’une plaie dans la tige, alors seules les cellules de la galle de la couronne résultante posséderont le gène cloné. Cela évidemment est de faible valeur pour le biotechnologiste. Au lieu de ça, l’introduction du nouveau gène dans chaque cellule de la plante est nécessaire. Il existe plusieurs solutions, la plus simple étant d’infecter non pas la plante mature mais plutôt la culture de cellules végétales ou de protoplastes en milieu liquide. Les cellules végétales et les protoplastes dont les parois cellulaires se sont formés peuvent être traités de la même manière que les micro-organismes: par exemple, ils peuvent être cultivées sur un milieu sélectif pour isoler les transformants. Une plante mature régénérée à partir de cellules transformées contiendra le gène cloné dans chaque cellule et passera le gène cloné à sa progéniture. cependant, La régénération d’une plante transformée ne peut se produire que si le vecteur Ti a été « désarmé » De sorte que les cellules transformées ne présentent pas de propriétés cancéreuses. Le désarmement est possible Parce que les gènes du cancer, qui sont tous dans l’ADN-T, ne sont pas nécessaires pour l’infection , L’infectiosité étant contrôlée principalement par la région de virulence du Ti plasmide. En fait, les seules parties de l’ADN-T qui sont impliquées dans l’infection sont deux séquences de répétitions de 25 pb trouvées aux frontières gauche et droite de la région L’ADN de la plante. Tout ADN placé entre ces deux séquences répétées sera traité comme « T-ADN » et transféré à la plante. Il est donc possible d’éliminer tous les gènes de cancers de l’ADN-T normal, et de les remplacer par un ensemble entièrement nouveau de gènes, Sans perturber le processus d’infection. Un certain nombre de vecteurs de clonage Ti désarmés sont maintenant disponibles, un exemple typique étant Le vecteur binaire pBIN19.
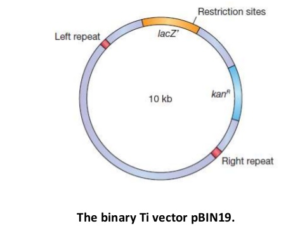
Les frontières de T-ADN gauche et droite Ce vecteur flanque une copie du gène lacZ ‘, contenant un certain nombre de sites de clonage, et un Gène de résistance à la kanamycine qui fonctionne après l’intégration des séquences Le chromosome végétal. Comme avec un vecteur navette de levure, les manipulations initialesQui entraînent l’insertion du gène à cloner dans pBIN19 sont réalisées dans E. coli, La molécule recombinante pBIN19 recombinante étant alors transférée à A. tumefaciens et De là dans la plante. Les cellules végétales transformées sont sélectionnées par étalement sur un milieu gélose Contenant de la kanamycine.
Le plasmide Ri :
Au fil des ans, il y a eu également un intérêt à développer des vecteurs de clonage basés sur le plasmide Ri d’Agrobacterium rhizogenes. Les plasmides Ri et Ti sont très semblables et la principale différence étant que le transfert de l’ADN-T d’un plasmide Ri à une plante a pour résultat non pas une galle de la couronne mais plutot la maladie des racines poilues, caractérisées par une racine très ramifiée. La possibilité de cultiver des racines transformées dans une culture liquide à haute densité a été exploré comme un moyen potentiel de produire des grandes quantités de protéines à partir de gènes clonés dans ces plantes.
Les limites du clonage avec des plasmides d’Agrobacterium :
Les plantes supérieures sont divisées en deux grandes catégories, les monocotylédones et les dicotylédones. Plusieurs facteurs se sont combinés pour rendre beaucoup plus facile le clonage de gènes chez les dicotylédones (la tomate, le tabac, la pomme de terre, les pois et les haricots) mais beaucoup plus difficile d’obtenir les mêmes résultats avec les monocotylédones. Cela a été frustrant parce les monocotylédones comprennent le (blé, L’orge, le riz et le maïs), qui sont les plantes de culture les plus importantes et donc les Cibles souhaitables pour les projets d’ingénierie génétique. La principale difficulté vient du fait que dans la nature l’A. tumefaciens et A. rhizogenes Infectent uniquement les plantes dicotylédones; Les monocotylédones sont en dehors de la gamme normale d’hôtes. Pendant quelque temps, on a pensé que cette barrière naturelle était insurmontable et que les Monocotylédones étaient totalement résistantes à la transformation avec des vecteurs Ti et Ri, mais finalement Des techniques artificielles pour réaliser le transfert d’ADN-T ont été imaginées. La transformation avec un vecteur d’Agrobacterium normalement Implique la régénération d’une plante intacte à partir d’un protoplaste, d’une cellule ou d’un calus transformés en Culture. La facilité avec laquelle une plante peut être régénérée dépend beaucoup des espèces particulières concernées et, encore une fois, les plantes les plus difficiles sont les monocotylédones. Les tentatives pour contourner ce problème se sont concentrées sur l’utilisation de bombardement biologique avec des microprojectiles pour introduire directement l’ADN plasmidique dans les embryons végétaux. Bien qu’il s’agisse d’une procédure de transformation assez violente, semblant trop dommageable pour les embryons, en réalité ces derniers continuent leur développement normal Pour produire des plantes matures. L’approche a été couronnée de succès avec le maïs Et plusieurs autres monocotylédones importantes.
Transfert direct de gènes:
Transfert direct de gènes dans le noyau:
Le transfert direct de gènes est basé sur l’observation, d’abord faite en 1984, qu’un plasmide bactérien super-enroulé, bien qu’il ne soit pas capable de se reproduire seul dans une cellule végétale, puisse être intégré par recombinaison dans l’un des chromosomes végétaux. L’événement de recombinaison est mal compris mais est presque certainement distinct des processus responsables de l’intégration d’ADN-T. Elle est également distincte de l’intégration chromosomique d’un vecteur de levure, car il n’existe aucune exigence pour une région de similarité entre le plasmide bactérien et l’ADN végétal. En fait, l’intégration semble se produire de façon aléatoire à n’importe quelle position dans n’importe lequel des chromosomes végétaux. Le transfert direct de gène utilise donc un ADN de plasmide super-enroulé, éventuellement un plasmide bactérien simple, dans lequel un marqueur sélectionnable approprié (par exemple, un gène de résistance à kanamycine) et le gène à cloner ont été insérés. La biolistique est fréquemment utilisée pour introduire l’ADN plasmidique dans des embryons végétaux, mais si l’espèce à cloner Peut être régénérée à partir de protoplastes ou des cellules uniques, alors d’autres stratégies, éventuellement plus efficaces que la biolistique, sont possibles. Un procédé consiste à remettre en suspension des protoplastes dans une solution visqueuse de polyéthylèneglycol, un composé polymère chargé négativement qui est censé précipiter l’ADN sur les surfaces des protoplastes et induire une absorption par endocytose (figure 7.16).

L’électroporation est également utilisée pour augmenter la fréquence de transformation. Après traitement, les protoplastes sont laissés pendant quelques jours dans une solution qui favorise la régénération des parois cellulaires. Les cellules sont ensuite étalées sur un milieu sélectif pour identifier les transformants et pour fournir des cultures de cals à partir desquelles des plantes intactes peuvent être cultivées (exactement comme décrit pour le système d’Agrobacterium).
Transfert de gènes dans le génome des chloroplastes :
Si la biolistique est utilisée pour introduire l’ADN dans un embryon végétal, certaines particules peuvent pénétrer un ou plusieurs des chloroplastes présents dans les cellules. Les chloroplastes contiennent leur propre génome, distinct et beaucoup plus court que les molécules d’ADN du noyau, et dans certaines circonstances, l’ADN plasmidique peut s’intégrer dans ce génome du chloroplaste. Contrairement à l’intégration de l’ADN dans les chromosomes nucléaires, l’intégration dans le génome des chloroplastes ne se produira pas de façon aléatoire. Au lieu de cela, l’ADN à cloner doit être flanqué de séquences similaires à la région du génome du chloroplaste dans laquelle l’ADN doit être inséré, de sorte que l’insertion peut avoir lieu par recombinaison homologue.
Chacune de ces séquences flanquantes doit avoir une longueur d’environ 500 pb. Un faible niveau de transformation du chloroplaste peut également être obtenu après l’administration d’ADN induite par PEG dans des protoplastes si le plasmide qui est prélevé porte ces séquences flanquantes. Une cellule végétale contient des dizaines de chloroplastes, et probablement seulement une par cellule devient transformée, de sorte que l’ADN inséré doit porter un marqueur sélectionnable tel que le gène de résistance à la kanamycine, et les embryons doivent être traités avec l’antibiotique pendant une période considérable pour s’assurer que Les génomes transformés se propagent dans la cellule. Bien que cela signifie que la transformation des chloroplastes est une méthode difficile à mettre en œuvre avec succès, elle pourrait devenir un adjuvant important aux méthodes plus traditionnelles pour obtenir des cultures OGM. Comme chaque cellule possède de nombreux chloroplastes, mais seulement un noyau, un gène inséré dans le génome du chloroplaste est susceptible d’être exprimé à un niveau supérieur à celui placé dans le noyau. Ceci est particulièrement important lorsque les plantes modifiées doivent être utilisées pour la production de protéines pharmaceutiques. Jusqu’à présent, l’approche a été la plus réussie avec le tabac, mais la transformation des chloroplastes a également été réalisée avec des cultures plus utiles comme le soja et le coton.
Utilissation des virus végétaux comme vecteurs de clonage :
Les versions modifiées des bactériophages λ et M13 sont des vecteurs de clonage importants pour E. coli. La plupart des plantes sont sujettes à une infection virale, est ce que les virus pourraient être utilisés pour cloner les gènes dans les plantes? Si elles le pouvaient, elles seraient beaucoup plus pratiques à utiliser que d’autres types de vecteurs, car avec de nombreux virus la transformation peut être obtenue simplement en frottant l’ADN du virus sur la surface d’une feuille. Le processus d’infection naturelle répand ensuite le virus dans toute la plante. Le potentiel des virus végétaux comme vecteurs de clonage a été exploré pendant plusieurs années, mais sans grand succès. Un problème est que la grande majorité des virus végétaux ont des génomes non d’ADN mais d’ARN. Les virus à ARN ne sont pas aussi utiles que les vecteurs de clonage potentiels car les manipulations avec l’ARN sont plus difficiles à réaliser. Seules deux classes de virus à ADN sont connues pour infecter des plantes supérieures, les caulimovirus et les geminivirus, et aucune d’entre elles n’est idéalement adaptée au clonage de gènes.
Les vecteurs à Caulimovirus :
Bien que l’une des premières expériences réussies d’ingénierie génétique végétale, en 1984, a utilisé un vecteur de caulimovirus pour cloner un nouveau gène dans des plantes de navet, deux difficultés générales avec ces virus ont limité leur utilité. La première est que la taille totale d’un génome de caulimovirus est, comme celle de e, contrainte par la nécessité de l’emballer dans sa couche de protéine. Même après la suppression de sections non essentielles du génome viral, la capacité de transporter l’ADN inséré est encore très limitée. Des recherches récentes ont montré qu’il serait possible de contourner ce problème en adoptant une stratégie de virus auxiliaire, semblable à celle utilisée avec les phagemides . Dans cette stratégie, le vecteur de clonage est un génome du virus de la mosaïque du chou-fleur (CaMV) qui manque de plusieurs des gènes essentiels, ce qui signifie qu’il peut transporter un gros segment d’ADN mais ne peut pas par lui-même causer une infection directe. Les plantes sont inoculées avec l’ADN vecteur avec un génome CaMV normal. Le génome viral normal fournit les gènes nécessaires pour que le vecteur de clonage soit emballé dans des protéines virales et se propage à travers la plante. Cette approche a un potentiel considérable, mais ne résout pas le deuxième problème, qui est la gamme extrêmement étroite de l’hôte de caulimovirus. Cela limite les expériences de clonage à quelques plantes seulement, principalement des champignons tels que les navets, les choux et les choux-fleurs. Les caulimovirus ont toutefois joué un rôle important dans le génie génétique comme source de promoteurs très actifs qui fonctionnent dans toutes les plantes et qui sont utilisés pour obtenir l’expression de gènes introduits par clonage de plasmide Ti ou transfert de gène direct.
Les vecteurs à Geminivirus:
Ceux-ci sont particulièrement intéressants parce que leurs hôtes naturels comprennent des plantes telles que le maïs et le blé, et ils pourraient donc être des vecteurs potentiels pour ces monocotylédones et d’autres. Mais les geminivirus ont présenté leur propre série de difficultés, un problème étant que pendant le cycle d’infection, les génomes de certains geminivirus subissent des réarrangements et des déletions, ce qui entraînerait un quelconque ADN supplémentaire qui a été inséré, un désavantage évident pour un vecteur de clonage. La recherche au fil des ans a abordé ces problèmes, et les geminivirus commencent à trouver des applications spécialisées dans le clonage de gènes de plantes. L’un d’entre eux est le séquençage génique induit par virus (VIGS), une technique utilisée pour étudier les fonctions des gènes de plantes individuelles. Cette méthode exploite l’un des mécanismes de défense naturelle que les plantes utilisent pour se protéger contre les attaques virales. Cette méthode, appelée ARN silencieux, entraîne une dégradation des ARNm viraux. Si l’un des ARN viraux est transcrit à partir d’un gène cloné contenu dans un génome de geminivirus, alors non seulement les transcrits viraux mais aussi les ARNm cellulaires dérivés de la copie du gène de la plante sont dégradés (figure 7.17). Le gène végétal devient donc silencieux et l’effet de son inactivation sur le phénotype de la plante peut être étudié.
Chapitre 4 : Vecteurs de clonage pour les eucaryotes: levures et autres champignons
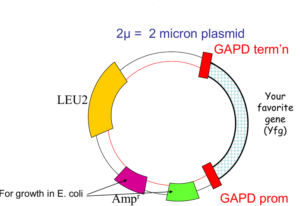
La levure Saccharomyces cerevisiae est l’un des organismes les plus importants dans la biotechnologie. Outre son rôle dans le brassage et la fabrication du pain, la levure a été utilisé comme un organisme hôte pour la production de produits pharmaceutiques importants à partir de gènes clonés. Dans un premier temps le développement des vecteurs de clonage de la levure, a été stimulée, par la découverte d’un plasmide qui est présent dans la plupart des souches de S. cerevisiae. Le 2 µ plasmide, comme on l’appelle, est l’un des rares plasmides trouvés dans les cellules eucaryotes.
Les marqueurs sélectionnables pour le plasmide 2 µm:
Le plasmide de 2 µm est un excellent élément pour un vecteur de clonage. Il a une taille de 6 kb, ce qui est idéal pour un vecteur et existe dans la cellule de levure à un nombre de copies compris entre 70 et 200. La réplication utilise une origine de plasmide, plusieurs enzymes fournies par la cellule hôte et les protéines codées Par les gènes REP1 et REP2 portés par le plasmide. Cependant, tout n’est pas parfaitement simple dans l’utilisation du plasmide de 2 μm comme vecteur de clonage. Tout d’abord, il y a la question d’un marqueur sélectionnable. Certains vecteurs de clonage de levure portent des gènes conférant une résistance aux inhibiteurs tels que le méthotrexate et le cuivre, mais la plupart des vecteurs de levure populaires utilisent un type de sélection radicalement différent.
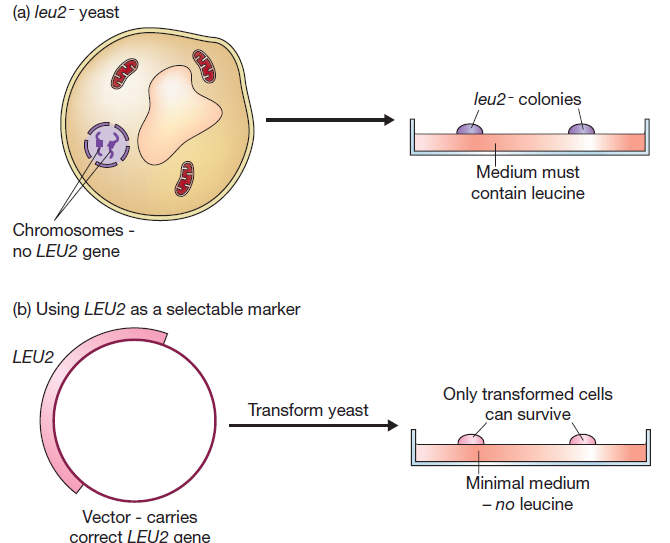
En pratique, on utilise un gène de levure normal, généralement celui qui code pour une enzyme impliquée dans la biosynthèse des acides aminés. Un exemple est le gène LEU2, qui code pour la b-isopropyl-malate déshydrogénase, l’une des enzymes impliquées dans la conversion de l’acide pyruvique en leucine. Afin d’utiliser le LEU2 comme marqueur de sélection, un type particulier d’organisme hôte est nécessaire. L’hôte doit être un mutant auxotrophique qui possède un gène LEU2 non fonctionnel. Une telle levure leu2- est incapable de synthétiser la leucine et ne peut survivre que si cet acide aminé est fourni comme nutriment dans le milieu de croissance . La sélection est possible parce que les transformants contiennent une copie du gène LEU2 supportée par un plasmide et sont ainsi capables de se développer en l’absence de l’acide aminé. Dans une expérience de clonage, les cellules sont étalées sur un milieu minimal, qui ne contient pas d’acides aminés ajoutés. Seules les cellules transformées sont capables de survivre et de former des colonies.
Les vecteurs basés sur le plasmide 2 µm – les plasmides épisomiques :
Les vecteurs dérivés du plasmide de 2 μm sont appelés plasmides épisomiques de levure (YEps=yeast episomal plasmids). Quelques YEps contiennent le plasmide de 2 µm en entier, d’autres incluent juste l’origine de réplication de 2 μm. Un exemple de ce dernier type est YEp13. YEp13 illustre plusieurs caractéristiques générales de vecteurs de clonage de levure. Tout d’abord, il s’agit d’un vecteur navette. Outre l’origine de replication de 2 μm et le gène LEU2 sélectionnable, YEp13 inclut également la séquence entière de pBR322, et peut donc se répliquer et être sélectionné pour à la fois dans la levure et dans E. coli.
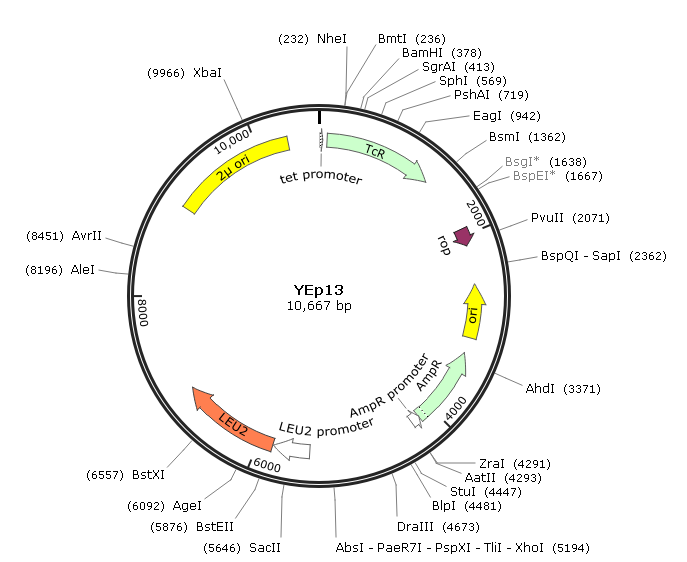
Il ya plusieurs lignes de raisonnement derrière l’utilisation des vecteurs navette. L’une est qu’il pourrait être difficile de récupérer la molécule d’ADN recombinant d’une colonie de levure transformée. Ce n’est pas un tel problème avec YEps, qui sont présents dans les cellules de levure principalement sous forme de plasmides, mais avec d’autres vecteurs de levure, qui peuvent s’intégrer dans un des chromosomes de levure, la purification pourrait être impossible. Ceci est un inconvénient car, dans de nombreuses expériences de clonage, la purification de l’ADN recombinant est essentielle pour que la construction correcte soit identifiée par exemple par séquençage d’ADN. La procédure standard lors du clonage dans la levure est donc d’effectuer l’expérience de clonage initiale avec E. coli et de sélectionner des recombinants dans cet organisme. Les plasmides recombinants peuvent ensuite être purifiés, caractérisés et la molécule correcte Introduite dans la levure.
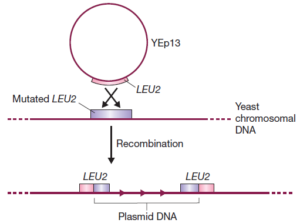
Un YEp peut s’insérer dans l’ADN chromosomique de levure
Le mot «épisomale» indique qu’un YEp peut se répliquer en tant que plasmide indépendant, mais implique également l’intégration dans l’un des chromosomes de levure (voir la définition de «épisome»). L’intégration se produit parce que le gène porté sur le vecteur comme marqueur de sélection est très similaire à la version mutante du gène présent dans l’ADN chromosomique de levure. Avec YEp13, par exemple, une recombinaison homologue peut se produire entre le gène du plasmide LEU2 et le gène LEU2 du mutant de levure, provoquant l’insertion du plasmide entier dans l’un des chromosomes de levure. le Plasmide peut rester intégré, ou un événement de recombinaison ultérieur peut l’excisé de nouveau.
 Autres types de vecteur de clonage de levure :
Autres types de vecteur de clonage de levure :
En plus des YEps , il existe plusieurs autres types de vecteurs de clonage à utiliser avec S.cerevisiae. Les deux plus importants sont les suivantes:
A) Les plasmides intégrés dans la levure (YIps = Yeast integrative plasmids ) sont essentiellement des plasmides bactériens portant un gène de levure. Un exemple est YIp5, qui est le pBR322 avec un gène URA3 inséré.
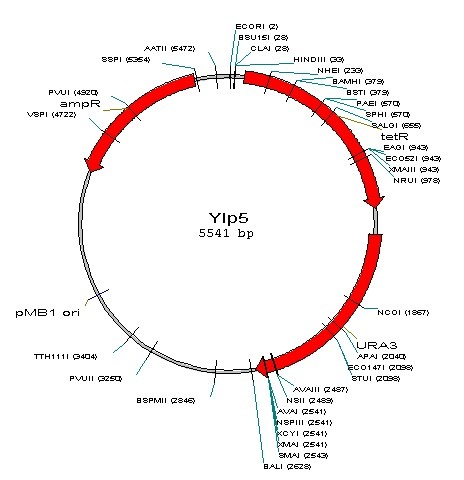
Ce gène code pour l’orotidine-5′-phosphate décarboxylase (une enzyme qui catalyse une des étapes de la voie de biosynthèse pour les nucleotides pyrimidiques) et est utilisée comme marqueur sélectionnable exactement de la même manière que LEU2. Un YIp ne peut pas se répliquer en tant que plasmide car il ne contient aucune partie du plasmide 2 µm et dépend plutôt pour sa survie de son intégration dans l’ADN chromosomique de levure. L’intégration se produit juste comme décrit pour un YEp.
B) Plasmides réplicatifs de levure (YRps=Yeast replicative plasmids )
Ils sont capables de se multiplier en tant que plasmides indépendants parce qu’ils portent une séquence d’ADN chromosomique qui comprend une origine de réplication. Les origines de réplication sont connues pour être situées très près de plusieurs des gènes de levure, comprenant un ou deux qui peuvent être utilisés comme marqueurs sélectionnables. YRp7 est un exemple de plasmide réplicatif. Il est composé de pBR322 plus le gène de levure TRP1. Ce gène, impliqué dans la biosynthèse du tryptophane, est situé adjacent à une origine chromosomique de réplication.
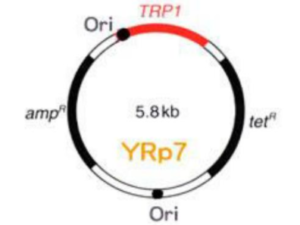
Le fragment d’ADN de levure présent dans YRp7 contient TRP1 et l’origine de réplication. Trois facteurs entrent en jeu pour décider quel type de vecteur de levure est le plus approprié pour une expérience de clonage particulière:
La première est la fréquence de transformation : une mesure du nombre de transformants qui peuvent être obtenus par microgramme de plasmidique. Une fréquence de transformation élevée est nécessaire si un grand nombre de recombinants sont nécessaires, ou si l’ADN de départ est insuffisant. YEps ont le plus haut fréquence de transformation, fournissant entre 10 000 et 100 000 cellules transformées Par µg. YRps sont également assez productifs, donnant entre 1000 et 10 000 transformants par µg, mais un YIp donne moins de 1000 transformants par µg et seulement 1-10 à moins que des procédures spéciales sont utilisées. La faible fréquence de transformation d’un YIp reflète que l’événement d’intégration chromosomique (qui est assez rare) est nécessaire avant que le vecteur puisse être conservé dans une cellule de levure.
Le deuxième facteur important est le nombre de copies. YEps et YRps ont le nombre de copie le plus élevé, 20-50 et 5-100, respectivement. En revanche, un YIp est habituellement présent à une seule copie par cellule. Ces chiffres sont importants si l’objectif est d’obtenir des protéines par le gène cloné, plus il y a de copies du gène, plus élevé sera le rendement attendu du produit protéique.
Alors pourquoi voudrait-on utiliser un YIp? La réponse est que YIps produit des recombinants très stables, comme la perte d’un YIp qui est devenu intégré dans un chromosome ne se produit qu’à une très faible fréquence. En revanche, les recombinants YRp sont extrêmement instables,les plasmides tendant à se rassembler dans la cellule mère lorsqu’une cellule fille se déclenche, la cellule fille n’est pas recombinante. Les recombinants YEp souffrent de problèmes similaires, bien qu’une meilleure compréhension de la biologie du plasmide de 2 µm ait permis de développer des YEps plus stable ces dernières années. Néanmoins, un YIp est le vecteur de choix si les besoins de l’expérience exigent que les cellules de levure recombinantes retiennent le gène cloné pendant de longues périodes en culture.
Les chromosomes artificiels peuvent être utilisés pour cloner de longs morceaux d’ADN dans des levures :
Le dernier type de vecteur de clonage de levure à considérer est le chromosome artificiel de levure (YAC), qui présente une approche totalement différente au clonage de gène. Le développement des YAC a été un spin-off de la recherche fondamentale sur la structure des chromosomes eucaryotes, le travail qui a identifié les composants clés d’un chromosome comme étant:
– le centromère, qui est nécessaire pour que le chromosome soit réparti correctement aux cellules filles pendant la division cellulaire;
– deux télomères, les structures aux extrémités d’un chromosome qui sont nécessaires pour que les extrémités soient correctement reproduites et qui empêchent également le chromosome d’être grignoté par les exonucléases;
– les origines de la réplication, qui sont les positions le long du chromosome à l’origine de la réplication de l’ADN, similaire à l’origine de la réplication d’un plasmide.
Une fois que la structure chromosomique a été définie de cette manière, la possibilité est apparue que les composants individuels puissent être isolés par des techniques d’ADN recombinant, puis rejoints ensemble dans le tube à essai, créant un chromosome artificiel. Comme les molécules d’ADN présentes dans les chromosomes naturels de levure ont plusieurs centaines de kilobases de longueur, il serait possible avec un chromosome artificiel de cloner de longs morceaux d’ADN.
La structure et l’utilisation d’un vecteur YAC
Plusieurs vecteurs YAC ont été développés mais chacun est construit selon le même schémas, le pYAC3 étant un exemple typique. À première vue, pYAC3 ne ressemble pas beaucoup à un chromosome artificiel mais en examinant de plus près ses caractéristiques uniques apparaissent. pYAC3 est essentiellement un plasmide pBR322 dans lequel un certain nombre de gènes de levure ont été insérés. Deux de ces gènes, URA3 et TRP1, ont déjà été rencontrés comme marqueurs sélectionnables pour YIp5 et YRp7, respectivement. Comme dans YRp7, le fragment d’ADN qui porte TRP1 contient également une origine de replication, mais dans pYAC3 ce fragment est étendu encore plus pour inclure la séquence appelée CEN4, qui est l’ADN provenant de la région centromère du chromosome 4. Le fragment (TRP1-origine- CEN4) contient donc deux des trois composants du chromosome artificiel. Le troisième composant, les télomères, est fourni par les deux séquences appelées TEL. Ce ne sont pas des séquences de télomères complètes, mais une fois à l’intérieur du noyau de levure, ils agissent comme des séquences d’ensemencement sur lesquelles les télomères seront construits. Il reste juste une autre partie de pYAC3 qui n’a pas été mentionnée: SUP4, qui est le marqueur sélectionnable dans lequel le nouvel ADN est inséré pendant l’expérience de clonage.
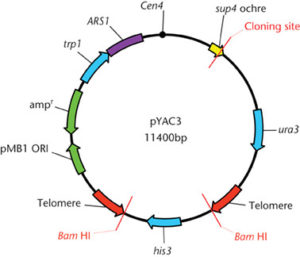
La stratégie de clonage avec pYAC3 est la suivante. Le vecteur est d’abord restreint avec une combinaison de BamHI et SnaBI, coupant la molécule en trois fragments. Le fragment flanqué de sites BamHI est jeté, laissant deux bras, chacun limité par une séquence TEL et un site SnaBI. L’ADN à cloner, qui doit avoir des extrémités émoussées (SnaBI est un cutter d’extrémité émoussée, reconnaissant la séquence TACGTA), est ligaturé entre les deux bras, produisant le chromosome artificiel.
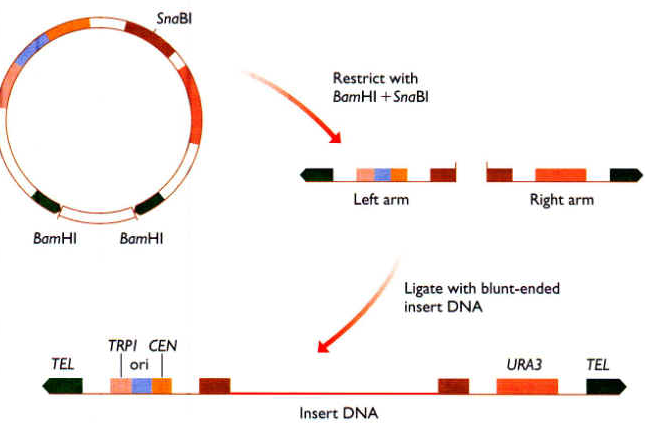
La transformation du protoplaste est alors utilisée pour introduire le chromosome artificiel dans S. cerevisiae. La souche de levure qui est utilisée est un double mutant auxotrope, trp1-ura3-, qui est converti en trp1 + ura3 + par les deux marqueurs sur le chromosome artificiel. Les transformants sont donc sélectionnés par culture sur milieu minimal, sur lequel seules des cellules contenant un chromosome artificiel correctement construit sont capables de se développer. Toute cellule transformée avec un chromosome artificiel incorrect, contenant deux bras droits ou deux bras gauches plutôt qu’un de chacun, n’est pas capable de croître sur un milieu minimal car l’un des marqueurs est absent. La présence de l’ADN d’insertion dans le vecteur peut être vérifiée en testant l’inactivation par insertion de SUP4, qui est réalisée par un simple test de couleur: les colonies blanches sont des recombinants, les colonies rouges ne le sont pas.
Utilisations des vecteurs YAC
Le stimulus initial dans la conception de chromosomes artificiels est venu de généticiens de levure qui voulait les utiliser pour étudier divers aspects de la structure et le comportement des chromosomes. Par exemple pour examiner la ségrégation des chromosomes lors de la méiose. Ces expériences ont établi que les chromosomes artificiels sont stables pendant la propagation dans des cellules de levure et ont soulevé la possibilité qu’ils pourraient être utilisés comme vecteurs pour des gènes qui sont trop longs pour être clones comme un seul fragment dans un vecteur d’E. coli. Plusieurs gènes de mammifères importants ont une longueur supérieure à 100 kb (par exemple, le gène de la mucoviscidose humaine est de 250 kb) qui est au-delà de la capacité de tous les systèmes de clonage d’E. Coli alors qu’ils sont dans la gamme d’un vecteur YAC. Les chromosomes artificiels de levure ont donc ouvert la voie à des études sur les fonctions et les modes d’expression de gènes qui avaient auparavant été intraitable à l’analyse par des techniques d’ADN recombinant. Une nouvelle dimension de ces expériences a été fournie par la découverte que, dans certaines circonstances, les YAC peuvent être propagés dans des cellules de mammifères, ce qui permet d’effectuer l’analyse fonctionnelle dans l’organisme dans lequel le gène réside normalement. Les chromosomes artificiels de levure sont également importants dans la production de banques de gènes. Rappelons qu’avec des fragments de 300 kb, la taille d’insert maximale pour le vecteur E. coli de capacité maximale, environ 30 000 clones sont nécessaires pour une banque de gènes humains. Cependant, les vecteurs YAC sont utilisés couramment pour cloner des fragments de 600 kb, et des types spéciaux sont capables de traiter l’ADN jusqu’à 1400 kb de longueur, ce dernier portant la taille d’une banque de gènes humains à seulement 6500 clones. Malheureusement, ces «méga-YAC» se heurtent à des problèmes de stabilité de l’insert, l’ADN cloné étant parfois réarrangé par recombinaison intramoléculaire. Néanmoins, les YAC ont été d’une immense valeur pour fournir de longues pièces d’ADN cloné pour une utilisation dans des projets de séquençage d’ADN à grande échelle.
Vecteurs pour d’autres levures et champignons :
Des vecteurs de clonage pour d’autres espèces de levures et de champignons sont nécessaires pour des études de base de la biologie moléculaire de ces organismes et pour étendre les utilisations possibles de levures et de champignons en biotechnologie. Les plasmides épisomiques basés sur le plasmide de 2 µm de S. cerevisiae sont capables de se répliquer dans quelques autres types de levures, mais la gamme des espèces n’est pas assez large pour que les 2 vecteurs µm soient de valeur générale. Dans tous les cas, les exigences de la biotechnologie sont mieux servies par des plasmides intégrateurs, équivalents à YIps, car ils fournissent des recombinants stables qui peuvent être cultivés pendant de longues périodes dans des bioréacteurs. Des vecteurs intégrateurs efficaces sont maintenant disponibles pour un certain nombre d’espèces, y compris des levures telles que Pichia pastoris et Kluveromyces lactis, et les champignons filamenteux tels que Aspergillus nidulans et Neurospora crassa.
Chapitre 3 : la remise de l’ADN cloné dans la cellule
Mettre l’ADN dans les cellules
Après que la séquence d’ADN à cloner a été joint au vecteur approprié, l’hybride doit être transféré dans des cellules pour l’amplification biologique. Le phénomène de la transformation des pneumocoques par l’ADN est connue depuis 1944. Une fois les propriétés génétiques souhaitables d’ E.coli ont été réalisées, elle aussi a été testée dans des expériences de transformation. Elles ont été infructueuses pendant de nombreuses années. Contre toute attente, un procédé de transformation d’E. coli a été découvert.

La Réintroduction d’une molécule d’ADN contenant un réplicon dans une cellule permet une amplification biologique de cette cellule à plus de 10 exposant 12. En un seul jour, une seule molécule peut être amplifiée à des quantités suffisantes pour des expériences physiques. La clé des protocoles de transformation initiale de E. coli a été le traitement des cellules avec des ions calcium ou de rubidium pour les rendre compétentes pour l’absorption du plasmide ou de l’ADN du phage. Le terme transformation avec de l’ADN du phage qui donne alors une cellule infectée est une transfection, et ce terme est aussi utilisé pour l’infection des cellules supérieurs avec l’ADN non viral. La levure peut être transfecté après un traitement qui comprend l’incubation, en présence d’ions lithium. Certains types de cellules de mammifère, des cellules L de souris, par exemple, peuvent être transfectés simplement en les arrosant avec un mélange de l’ADN et du phosphate de calcium cristaux. Ici, le mécanisme de transfection semble être l’absorption du complexe ADN-phosphate de calcium.
La Microinjection :
l’injection manuelle directe de petites quantités d’ADN dans les cellules a été très utile pour l’étude des fragments d’ADN clonés, car elle élimine la nécessité d’un réplicon ou un gène eucaryote sélectionnable. La microinjection dans les ovocytes de la grenouille Xenopus laevis a donné beaucoup d’information et la micro-injection dans des cellules de mammifère en culture est également possible. ADN injecté dans les cellules de Xenopus est transcrit pendant de nombreuses heures et traduit en quantités facilement détectables de protéine. En conséquence un segment d’ADN peut être clones sur un plasmide tel que pBR322, manipulé in vitro et injectée dans les cellules pour examiner ses nouvelles propriétés biologiques. La microinjection est également possible dans un embryon de souris fertilisés. L’embryon peut alors être réimplanté à une souris pour se développer. Étant donné que les fragments d’ADN injecté se recombinent dans le chromosome, les souris seront transfectées par l’ADN. Pour que toutes les cellules de souris transfectée possèdent le même patrimoine génétique le fragment injecté doit recombiner dans une cellule de lignée germinale. Une telle souris est peu susceptible d’être génétiquement homogène car probablement les fragments similaires n’ont probablement pas intégrés dans les cellules somatiques. La progéniture d’une telle souris cependant peut être génétiquement homogène, et celle-ci peut être étudiée .

L’ Electroporation :
Une autre méthode générale pour incorporer de l’ADN dans les cellules est l’électroporation. Les cellules sont soumises à un champ électrique brève mais intense. Ce-ci crée de petits trous dans leurs membranes et pour une courte durée les ADN présents dans la solution peuvent être absornéspar les cellules
Clonage à partir d’ARN :
Bien que l’ADN peut être extrait à partir des cellules et utilisé dans les étapes de clonage, parfois l’ARN est un meilleur choix de départ pour le clonage. Non seulement les séquences intermédiaires peuvent être manquantes à partir d’ARNm, mais souvent l’ARNm extrait à partir de certains tissus est fortement enrichi pour les séquences de gènes spécifiques. Extraction de l’ARN à partir des cellules donne une prépondérance d’ ARN ribosomal. L’ARN messager peut facilement être séparé de cet ARN ribosomal puisque la plupart des ARN messager de la plupart des organismes supérieurs contiennet une queue poly-A à l’extrémité 3 ‘. Cette queue peut être utilisé pour l’isolement en faisant passer une fraction brute de l’ARN cellulaire à travers une colonne de cellulose dans laquelle poly-dT a été lié. À des concentrations élevées en sel, les poly-dT qui sont reliées à la colonne et les queues poly-A des molécules qui sont lieés aux ARN messagers de la colonne. Les molécules d’ARN ribosomique circulent librement à travers la colonne. Les ARN messagers sont sont libérés par abaissement de la concentration en sel de manière à affaiblir le poly-dT hybrides. Une telle étape de purification fournit souvent un plusieurs-centuple d’enrichissement pour l’ARN messager. L’effort nécessaire pour cloner le gène spécifique est souvent fortement réduite par l’utilisation de cette procédure en conjonction avec le choix d’un tissu particulier à un développement particulier. L’ARN simple brin obtenu par les étapes décrites ci-dessus ne peut pas être directement clonés. Ou bien l’ARN peut être converti en ADN par l’intermédiaire d’un brin complémentaire, l’ADNc ou l’ARN peut être utilisé pour faciliter l’identification d’un clone contenant la séquence d’ADN complémentaire. À générer une copie d’ADNc du messager poly-A contenant, plusieurs étapes sont effectuées . Tout d’abord, une amorce poly-dT est hybridée à l’ARN messager et la transcriptase inverse est utilisé pour allonger l’apprêt et produire une copie de l’ADN. Cette enzyme, qui se trouve à l’intérieur du virus libre des particules de certains virus animaux, la synthèse de l’ADN en utilisant une matrice d’ARN. A ce stade, la séquence existe sous la forme d’un hybride ARN-ADN. Ceci se converti en un duplex d’ADN par incubation simultanée avec la RNAse H, qui coupe le brin d’ARN dans un duplex ARN-ADN et ADN polymérase pol I supprime le reste de l’ARN par translation de coupure. Enfin, pour rendre les extrémités du duplex d’ADN parfaitement émoussé, l’ADN polymerase T4 pol I est ajouté. L’ADN double brin ainsi obtenu peut ensuite être cloné par des méthodes déjà discutées.

Hybridation de Plaque et de Colonie pour l’identification de Clone :
De nombreuses techniques ont été mises au point pour la détection des clones souhaités. Un des plus simples est la sélection génétique (Fig. 9.13). Le plus souvent cette simple avenue ne sont pas disponibles, mais on possède une séquence apparentée. Parfois, cette séquence apparentée provient d’un gène analogue au gène désiré, mais à partir d’un organisme différent. D’autres fois, des séquences d’acides aminés sont connues ou bien une approximation peut être faite quant à la la séquence des acides aminés. A partir de ces séquences, les séquences d’ADN peuvent être
Une variété de techniques de criblage direct existent pour la détection d’un gène cloné. Dans ces approches,le clonage de l’ADN étranger dans un vecteur lambda est pratique parce que le phage peut accueillir des fragments insérés de grande dimension et un grand nombre de phage lambda peuvent être criblés sur une seule boite de petri . La collection de phage candidat est appelé une banque lambda ou d’une bibliothèque.
Il est étalé sur des milliers de plaques par plaque. Ensuite, une copie de la réplique de
les plaques de phage est réalisé en appuyant sur un papier filtre sur la plaque. Après
le papier est enlevé, il est immergé dans une solution alcaline. Par ces étapes l’ADN
est dénaturé et fixé sur le papier. L’ARN marqué radioactivement puis
ou de l’ADN, appelé une sonde est hybridée à un ADN quelconque complémentaire
séquences en images de plaque sur le papier. La sonde contient la connue
séquence dérivée d’un clone isolé au préalable ou d’une séquence déduite
à partir des informations de séquence amino-acide. L’emplacement des zones
avec sonde liée sont ensuite déterminées par autoradiographie, et viable
phagique contenant l’insert désiré peut être isolé à partir du composé
la position sur la plaque originale. techniques analogues existent pour le dépistage
les colonies contenant les plasmides.
Marcher le long d’une Chromosome cloner un gène
Une autre méthode de clonage d’un gène désiré est la marche. Supposons qu’un hasard
clone isolé a été montré pour se situer à l’intérieur de plusieurs centaines
mille bases du segment d’ADN que nous souhaitons cloner. Bien sûr,
démontrant un tel fait est pas banal en soi. Dans le cas du fruit
mouche Drosophila melanogaster, cependant, la démonstration est simple.
Comme expliqué précédemment, les chromosomes polytènes dans les cellules de plusieurs
tissus de Drosophila possèdent des bandes caractéristiques qui servent de chromoso-
(une)
a fragments b c d clonées
une
b
c
(B)
ré
Composite carte de restriction du segment chromosomique
Restriction enzyme
des sites de clivage
La figure 9.14 (a) Un gel représentant des fragments d’ADN résultant d’une restriction
digestion enzymatique d’un ensemble de quatre séquences d’ADN se chevauchant partiellement. (B) Le
quatre ADN et leur composite.
284 génie génétique et l’ADN recombinant
repères mal. Par conséquent, l’ADN impliquée dans des réarrangements chromosomiques
ou suppressions peuvent être facilement observés. En outre ADN hautement radioactifs
provenant d’un segment clone de drosophile chromosomique peut être
hybridées in situ à drosophile chromosomes polytènes. La position à
que les fragments se hybride peuvent alors être déterminées par autoradiographie.
Par conséquent, la position chromosomique d’un fragment peut être clone
déterminé. Si elle se trouve à proximité d’un gène d’intérêt, la marche peut être dans l’ordre.
Une chromosomique à pied de cloner un gène spécifique commence par une restriction
carte de la pièce d’ADN clone. Le terminal de droite et de gauche-end
les fragments de restriction sont utilisées en tant que sondes d’une banque lambda. Plusieurs
Les clones sont choisis qui hybrident le fragment de droite et de ne pas
le fragment de gauche. Ensuite, les cartes de restriction de ces clones sont fabriqués
et encore les fragments de droite et de gauche sont utilisés pour trouver de nouveaux clones
qui hybrident uniquement aux fragments de droite (Fig. 9.14). successif
lambda transduction phage qui sont identifiés permis marche du
à droite, et lorsqu’une distance suffisante a été couverte, une hybridation in situ
permet de déterminer dans quelle direction sur l’ADN clone
correspond
Google Traduction pour les entreprises :Google Kit du traducteurGadget TraductionOutil d’aide à l’export
Google Traduction pour les entreprises :Google Kit du traducteurGadget TraductionOutil d’aide à l’export
Chapitre 2 : les vecteurs
Vecteurs: la Sélection et réplication autonome de l’ ADN :
Le clonage d’un morceau d’ADN exige qu’il soit répliqué quand il est remis dans les cellules. D’où l’ADN à cloner doit lui-même être une unité répliquante indépendante, un réplicon ou doit être joint à un réplicon. De plus, étant donné que l’efficacité de l’introduction de l’ADN dans des cellules est bien inférieur à 100%, les cellules qui ont absorbé l’ADN et on dit qu’elles ont été transformées, doivent être facilement identifiables. En effet, étant donné que seulement une bactérie cellule sur 105 est transformée, les sélections doivent généralement être incluses pour autoriser uniquement les cellules transformées de croître. Les vecteurs doivent remplir les deux conditions décrites ci-dessus, la réplication dans la cellule hôte et la sélection des cellules ayant reçu l’ADN transformant. Comme mentionné précédemment deux principaux types de vecteurs sont utilisés : des plasmides et des phages. Les plasmides contiennent des réplicons bactériens qui peuvent coexister avec l’ADN cellulaire normal et au moins un gène sélectionnable. Habituellement c’est un gène conférant une résistance à une antibiotique. Les phages contiennent bien sûr des gènes pour la réplication de leur ADN. Comme l’ADN emballé dans une enveloppe de phage peut pénétrer dans les cellules de manière efficace des gènes sélectionnables sur les phages habituellement ne sont pas nécessaires.
Vecteur plasmidique :
La plupart des plasmides sont de petits cercles qui contiennent les éléments nécessaires pour la réplication de l’ADN, un ou deux gènes de résistance aux médicaments et une région d’ADN dans laquelle l’ADN étranger peut être inséré sans endommager les fonctions essentielles du plasmide.
Un plasmide largement utilisé, pBR322, porte des gènes codant pour la résistance à la tetracycline et β-lactamase celle-ci confère une résistance à la pénicilline et aux analogues apparentés en clivant les médicaments dans le cycle lactame, ce qui les rend biologiquement inactifs. Des gènes conférant une résistance au chloramphénicol, à la tétracycline et kanamycine sont d’autres marqueurs de résistance aux médicaments couramment sélectionnables portés sur des plasmides.
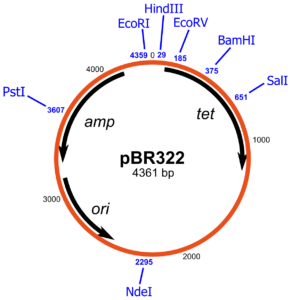
Un élément utile à avoir sur les plasmides est une origine de réplication de l’ADN à partir d’un phage simple brin. Quand une telle origine est activée par l’infection de tel phage, la cellule synthétise des quantités importantes d’un seul brin de l’ADN du plasmide. Ceci facilite le séquençage de l’ADN. Dans une expérience typique de clonage un plasmide est coupé dans une région non essentielle par une enzyme de restriction par exemple EcoRI et l’ADN monocaténaire étranger, coupé par EcoRI, est ajouté et les extrémités hybrides sont soudées ensemble. Seule une petite fraction des plasmides, soumis à ce traitement contiendra l’ADN inséré. La plupart auront recircularisé sans insertion d’ADN étranger. Comment les bactéries transformées c.à.d ceux dont les plasmides contiennent l’ADN inséré peuvent être distinguées des autres (ceux dont le plasmide reste sans ADN inséré)? Bien entendu dans certaines conditions une sélection génétique peut être utilisée pour activer uniquement la croissance des éléments transformés avec le fragment souhaité d’ADN inséré. Le plus souvent cela n’est pas possible et il devient nécessaire d’identifier des candidats qui contiennent l’ADN inséré.Une méthode pour identifier des candidats repose sur l’inactivation par l’ insertion d’un gène de résistance aux médicaments.
Par exemple, dans la résistance à l’ampicilline, le gène de la résistance sur pBR322 possède un seul site de clivage du plasmide par l’enzyme de restriction PstI. Heureusement le clivage par PstI génère des extrémités collantes et l’ADN peuvent être facilement soudées à ce niveau, après quoi il désactive le gène de la résistance à l’ampicilline. Le gène de résistance à la tétracycline sur le plasmide demeure intact et peut être utilisé pour la sélection des cellules transformées par le plasmide recombinant. Les colonies résultantes peuvent être testées en repérant sur une paire de plaques, l’une contenant de l’ampicilline, et l’autre sans ampicilline. Seul les mutants ampicilline sensibles, tétracycline-résistants vont contenir l’ADN étranger dans le plasmide alors que les ampicilline résistantess possèdent des plasmides re-circularisés sans insertion de l’ADN étranger.
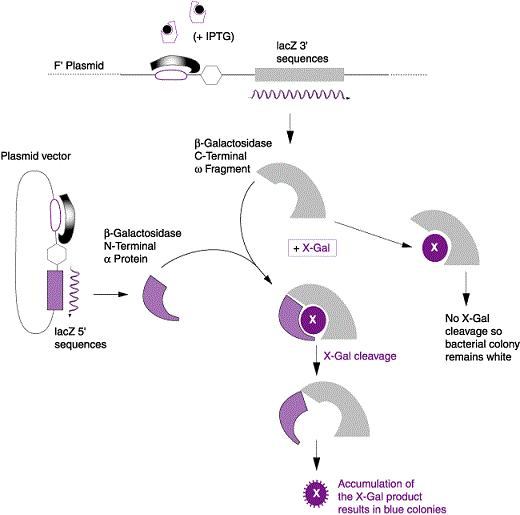
Une autre façon pour vérifier l’insertion d’ADN étranger, utilise le gène de ß-galactosidase. L’insertion d’ADN étranger dans le gène inactive l’enzyme qui peut être détectée en mettant les cellules transformées sur un milieu qui sélectionne la présence du plasmide et contient également des substrats de β-galactosidase qui produisent des colorants lorsqu’il hydrolyse ces substrats.
Un plasmide serait lourd s’il contenait la totalité des 3000 paires de bases du gène β galactosidase. Par conséquent seule une extrémité N-terminale du gène est placé sur le plasmide. Le reste de l’enzyme est codée par un segment inséré dans le chromosome des cellules hôtes. Les deux parties du gène synthétisent des domaines qui se lient entre eux pour donner l’enzyme actif. Ce phénomène inhabituel est appelé alpha-complémentation.
Des vecteurs de clonage sont conçus pour l’insertion d’ ADN étranger dans la partie N-terminale de la β-galactosidase. Une technique simple peut réduire considérablement la re-circularisation des molécules du vecteur sans insertion de l’ADN étranger. Si l’ADN du vecteur est traité par une phosphatase après coupure par l’enzyme de restriction, la re-circularisation devient impossible parce que l’extrémité 5′-PO4 requise pour l’ ADN ligase est absente. L’ADN étranger, cependant, contient une extrémité 5′-PO4 et par conséquent deux des quatre fragments d’ADN encadrant un fragment d’ADN étranger peuvent être soudés. Cet ADN est actif dans la transformation parce que les cellules réparent le pseudo restant à chaque extrémité du fragment inséré. Les vecteurs de clonage plasmides ou phages peuvent contenir un court tronçon de l’ADN contenant des sites de coupure unique pour plusieurs enzymes de restriction. Ces régions à liaisons multiples (polylinker) permettent le clivage par deux enzymes de sorte que les extrémités collantes résultantes ne sont pas auto-complémentaire. Donc le vecteur ne peut pas se re-circularisé et être resoudé sur lui-même mais quand un fragment d’ADN contenant les extrémités nécessaires et complémentaires aux extrémités du plasmide existe alors il peut le faire. Une génie génétique efficace nécessite que l’ADN plasmidique soit obtenu en grandes quantités. Certains plasmides maintiennent seulement trois ou quatre copies par cellule, tandis que d’autres plasmides ont 25 à 50 copies cellulaires. La plupart des plasmides ayant un nombre importants de copies peuvent être amplifiée du fait que le plasmide continue de se répliquer après que la synthèse protéique et la synthèse de l’ADN cellulaire ont cessé en raison d’une densité cellulaire élevée ou la présence d’inhibiteurs de la synthèse des protéines. Après amplification, une cellule contenant un tel plasmide peut contenir jusqu’à 3000 copies de plasmide.

La nature n’a pas livré des vecteurs plasmidiques prêt pour le génie génétique.Les vecteurs plasmidiques les plus utiles ont été eux-mêmes construits par génie génétique. Les matières de départ sont les plasmides R, ce sont des plasmides ou des éléments d’ADN à réplication autonome qui portent un ou plusieurs gènes de résistance aux médicaments. Les plasmides R sont la cause de graves problèmes médicaux, car diverses bactéries peuvent acquérir des plasmides R et ainsi devenir résistant aux médicaments normalement utilisés pour le traitement des infections . La conversion d’un plasmide R en un vecteur utile nécessite l’élimination de l’ADN étranger et l’élimination des multiples sites de clivage par des enzymes de restriction. Afin que l’ADN étranger puisse être cloné dans le plasmide, le plasmide doit posséder un seul site de clivage pour au moins une enzyme de restriction, et cela devrait être dans une région non essentielle. Dans la construction des vecteurs de clonage, des plasmides ont été digérés avec diverses enzymes de restriction. Le mélange résultant de fragments d’ADN a été hybridés ensemble par l’intermédiaire des extrémités auto-complémentaires, puis collées pour produire de nombreuses combinaisons de fragments possibles . Ces ADN ont été transférés dans des cellules. Seuls les nouveaux plasmides contenant au moins les segments d’ADN nécessaires à la réplication et à la résistance aux médicaments ont survécu et ont donné des colonies. Les plasmides souhaitables contenant uniquement les sites de clivage uniques pour des enzymes de restriction peuvent être identifiés par l’amplification et la purification de l’ADN suivie par des digestions test avec des enzymes de restriction suivi d’électrophorèse pour caractériser les Produits de la digestion. Le plasmide pBR322 possède une seule site de clivage pour plus de 20 enzymes de restriction. Parmi les plus communément utilisés sont Bam HI, Eco RI, Hind III, PstI, PvuII et Sali.
Un vecteur phagique pour les bactéries :
des vecteurs de phage et des vecteurs dérivés de phages sont utiles pour trois raisons:
-les phages peuvent transporter de plus grands fragments d’ADN insérés que des plasmides. Donc beaucoup moins de candidats transformés doivent être examinés pour trouver un clone désiré.
-L’efficacité d’infecter les cellules par l’ADN des phages reconditionnés est considérablement plus grande que l’efficacité de la transformation avec L’ADN de plasmide dans les cellules. Ceci est un facteur important quand un clone rare est recherché.
-Enfin, le phage lambda permet un procédé commode pour le criblage pour détecter le clone porteur du gène désiré. Cependant une fois qu’un segment désiré d’ADN a été cloné dans un phage la commodité de manipuler des plasmides, en partie en raison de leur petite taille, dicte que le segment soit sous-cloné dans un plasmide.
Vecteur de clonage basé sur le bactériophage M13 :
L’exigence la plus essentielle pour tout vecteur de clonage est qu’il ait un moyen de se répliquer dans la cellule hôte. Pour les vecteurs plasmidiques, cette exigence est facile à satisfaire, car des séquences d’ADN relativement courtes sont capables d’agir comme origines de réplication plasmidiques, et la plupart, sinon la totalité, des enzymes nécessaires à la réplication sont fournies par la cellule hôte. Des manipulations élaborées, telles que celles qui ont abouti à pBR322, sont donc possibles tant que la construction finale a une origine de réplication fonctionnelle et intacte. Avec des bactériophages tels que M13 et e, la situation en matière de réplication est plus complexe. Les molécules d’ADN phagique portent généralement plusieurs gènes essentiels à la réplication, y compris des gènes codant pour des composants de la couche protéique du phage et des enzymes réplicatives spécifiques de l’ADN phagique. La modification ou la suppression de l’un de ces gènes altèrent ou détruisent la capacité de réplication de la molécule résultante. Il y a donc beaucoup moins de liberté pour modifier les molécules d’ADN phagique, et généralement les vecteurs de clonage de phages ne sont que légèrement différents de la molécule parente.
Les problèmes de construction d’un vecteur de phages sont illustrés en considérant M13. Le génome M13 normal a une longueur de 6,4 kb, mais la plus grande partie de ce génome est occupée par dix gènes étroitement liés, chacun étant essentiel à la réplication du phage. Il n’y a qu’une seule séquence intergénique de 507 nucléotides dans laquelle un nouvel ADN pourrait être inséré sans perturber l’un de ces gènes, et cette région comprend l’origine de réplication qui doit elle-même rester intacte. Clairement, il n’y a qu’une possibilité limitée de modifier le génome M13.
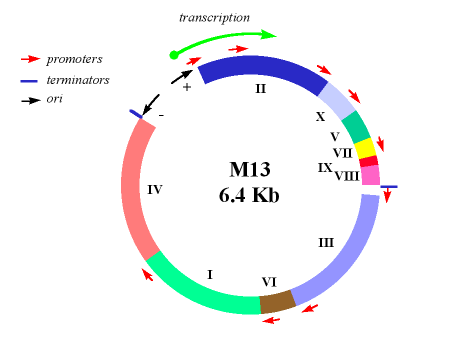
La première étape de construction d’un vecteur M13 consistait à introduire le gène lacZ ‘dans la séquence intergénique. Cela a donné M13mp1, qui forme des plaques bleues sur de l’agar X-gal.
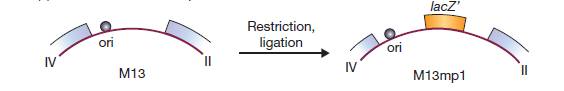
M13mp1 ne possède pas de sites de restriction uniques dans le gène lacZ ‘. Cependant, il contient l’hexanucléotide GGATTC près du début du gène. Un seul changement de nucléotide rendrait ce GAATTC, qui est un site EcoRI.
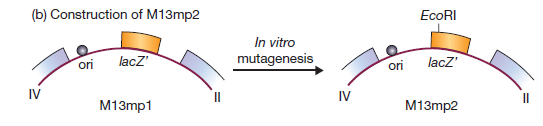
Cette altération a été réalisée en utilisant la mutagenèse in vitro, résultant en M13mp2
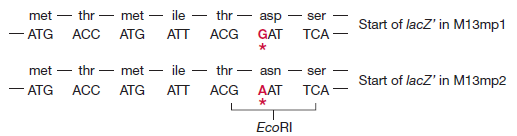
M13mp2 a un gène lacZ′ légèrement modifié (le sixième codon spécifie maintenant l’asparagine à la place de l’acide aspartique), mais l’enzyme β-galactosidase produite par les cellules infectées par M13mp2 est encore parfaitement fonctionnelle. L’étape suivante dans le développement des vecteurs M13 consistait à introduire des sites de restriction supplémentaires dans le gène lacZ’. Ceci a été réalisé en synthétisant dans le tube à essai un oligonucléotide court, appelé polylinker, qui consiste en une série de sites de restriction et a des extrémités collantes EcoRI.
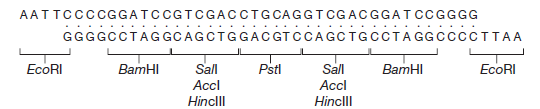
Ce linker multisite a été inséré dans le site EcoRI de M13mp2, pour donner M13mp7, un vecteur plus complexe avec quatre sites de clonage possibles (EcoRI, BamHI, Sali et PstI). Le polylinker est conçu pour ne pas perturber totalement le gène lacZ’: un cadre de lecture est maintenu dans tout le polylinker, et une enzyme β-galactosidase fonctionnelle bien que modifiée est encore produite. Les vecteurs M13 les plus sophistiqués ont des polylinkers plus complexes insérés dans le gène lacZ’. Un exemple est M13mp8, qui a la même série de sites de restriction que le plasmide pUC8 . Comme avec le vecteur plasmidique, un avantage de M13mp8 est sa capacité à prendre des fragments d’ADN avec deux extrémités cohésives différentes.
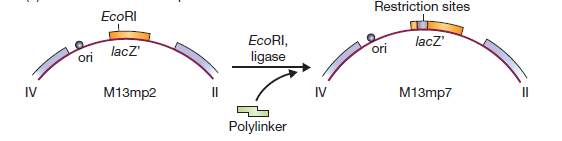
Phagemides (vecteurs hybrides formés de plasmides et M13) :
Bien que les vecteurs M13 soient très utiles pour la production de versions monocaténaires de gènes clonés, ils souffrent d’un inconvénient. Il y a une limite à la taille du fragment d’ADN qui peut être cloné avec un vecteur M13, avec 1500 pb comme la capacité maximale, bien que des fragments jusqu’à 3 kb ont parfois été cloné. Pour contourner ce problème, un certain nombre de vecteurs hybrides (« phagemides ») ont été développé en combinant une partie du génome M13 avec l’ADN plasmidique. Un exemple est fourni par pEMBL8, qui a été faite en transférant dans pUC8 un fragment de 1300 pb du génome M13.
Ce morceau d’ADN M13 contient la séquence signal reconnue par les enzymes qui convertissent le double brin normal de M13 en ADN simple brin avant la sécrétion de nouvelles particules phagiques. Cette séquence de signaux est toujours fonctionnelle même si elle est détachée du reste du génome de M13, les molécules pEMBL8 sont également converties en ADN simple brin et sécrété comme particules phagiques défectueuses.
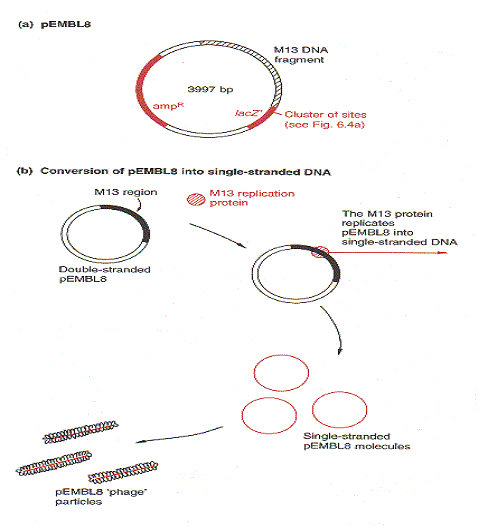
Tout ce qui est nécessaire, c’est que les cellules de E. coli utilisées en tant qu’hôtes pour une expérience de clonage pEMBL8 sont ensuite infectés avec M13 normal à agir comme un phage auxiliaire, fournissant les enzymes réplicatives nécessaires et les protéines de l’enveloppe du phage. pEMBL8, dérivé de pUC8, a les sites de clonage de polylinker dans le lacZ ‘ gène, de sorte que les plaques recombinantes peuvent être identifiées de manière standard sur de l’agar contenant X-gal. Avec pEMBL8, versions monocaténaires de fragments d’ADN clonés jusqu’à 10 kb en longueur peut être obtenue, en étendant considérablement la portée du système de clonage M13.
Vecteurs de clonage basés sur bactériophage λ :
Deux problèmes devaient être résolus avant que les vecteurs de clonage basés sur λ puissent être développés:
l) La molécule d’ADN de λ peut être augmentée seulement d’environ 5%, ce qui représente l’addition de seulement 3 kb de nouveaux ADN. Si la taille totale de la molécule est supérieure à 52 kb, alors elle ne pourra pas être empaquetée dans la structure de la tête e et les particules de phages infectieuses ne sont pas formées. Ceci limite sévèrement la taille d’un fragment d’ADN qui peut être inséré dans un vecteur λ non modifié.
2) Le génome λ est si grand qu’il a plus d’une séquence de reconnaissance pour pratiquement chaque endonucléase de restriction. La restriction ne peut pas être utilisée pour cliver la molécule λ normale de manière à permettre l’insertion d’un nouvel ADN, car la molécule serait coupée en plusieurs petits fragments qui seraient très peu susceptibles de reformer un génome λ viable lors de la religation. Au vu de ces difficultés, il est peut-être surprenant qu’une grande variété de vecteurs de clonage λ aient été développés, leur utilisation principale étant de cloner de gros morceaux d’ADN de 5 à 25 kb, beaucoup trop gros pour être manipulés par des vecteurs plasmidiques ou M13.
Des segments du génome λ peuvent être supprimés sans compromettre sa viabilité :
La voie à suivre pour le développement de vecteurs de clonage a été fournie par la découverte qu’un grand segment dans la région centrale de la molécule d’ADN peut être éliminé sans affecter la capacité du phage à infecter des cellules de E. coli. L’élimination de tout ou partie de cette région non essentielle, jusqu’à 15 kb. Cela signifie que jusqu’à 18 kb de nouvel ADN peuvent maintenant être ajoutés avant que le seuil de l’emballage ne soit atteint . Cette région « non-essentielle » contient en effet la plupart des gènes impliqués dans l’intégration et l’excision du prophage λ du chromosome de E. coli. Un génome de λ modifié par suppression est donc non lysogène et ne peut suivre que le cycle d’infection lytique. Ceci est en soi souhaitable pour un vecteur de clonage car cela signifie qu’une induction n’est pas nécessaire avant la formation de plaques .
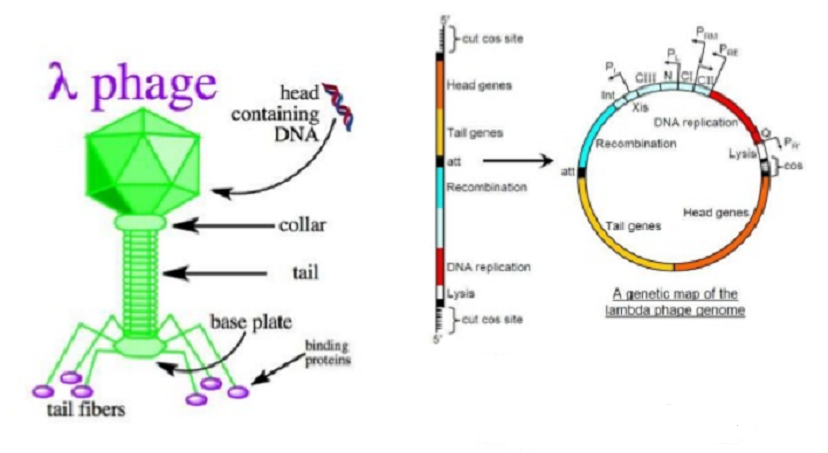
La sélection naturelle peut être utilisée pour isoler des λ modifiés dépourvus de certains sites de restriction :
Natural selection can be used to isolate modified * that lack certain restriction sites Even a deleted e genome, with the non-essential region removed, has multiple recognition sites for most restriction endonucleases. This is a problem that is often encountered when a new vector is being developed. If just one or two sites need to be removed, then the technique of in vitro mutagenesis (p. 200) can be used. For example, an EcoRI site, GAATTC, could be changed to GGATTC, which is not recognized by the enzyme. However, in vitro mutagenesis was in its infancy when the first e vectors were under development, and even today would not be an efficient means of changing more than a few sites in a single molecule. Instead, natural selection was used to provide strains of e that lack the unwanted restriction sites. Natural selection can be brought into play by using as a host an E. coli strain that produces EcoRI. Most e DNA molecules that invade the cell are destroyed
by this restriction endonuclease, but a few survive and produce plaques. These are mutant phages, from which one or more EcoRI sites have been lost spontaneously (Figure 6.11). Several cycles of infection will eventually result in e molecules that lack all or most of the EcoRI sites.
6.3.3 Insertion and replacement vectors
Once the problems posed by packaging constraints and by the multiple restriction sites had been solved, the way was open for the development of different types of λ-based cloning vectors. The first two classes of vector to be produced were λ insertion and λ replacement (or substitution) vectors.
λ Insertion vectors
With an insertion vector (Figure 6.12a), a large segment of the non-essential region has
been deleted, and the two arms ligated together. An insertion vector possesses at least
one unique restriction site into which new DNA can be inserted. The size of the DNA
fragment that an individual vector can carry depends, of course, on the extent to which
the non-essential region has been deleted. Two popular insertion vectors are:
l Egt10 (Figure 6.12b), which can carry up to 8 kb of new DNA, inserted into a
unique EcoRI site located in the cI gene. Insertional inactivation of this gene means
that recombinants are distinguished as clear rather than turbid plaques (p. 83).
λ replacement vectors.
A e replacement vector has two recognition sites for the restriction endonuclease used
for cloning. These sites flank a segment of DNA that is replaced by the DNA to be
cloned (Figure 6.13a). Often the replaceable fragment (or “stuffer fragment” in cloning
jargon) carries additional restriction sites that can be used to cut it up into small pieces,
so that its own re-insertion during a cloning experiment is very unlikely. Replacement
vectors are generally designed to carry larger pieces of DNA than insertion vectors can
handle. Recombinant selection is often on the basis of size, with non-recombinant vectors
being too small to be packaged into e phage heads (p. 84).
An example of a replacement vectors is:
l EEMBL4 (Figure 6.13b) can carry up to 20 kb of inserted DNA by replacing a
segment flanked by pairs of EcoRI, BamHI, and SalI sites. Any of these three
restriction endonucleases can be used to remove the stuffer fragment, so DNA
fragments with a variety of sticky ends can be cloned. Recombinant selection with
eEMBL4 can be on the basis of size, or can utilize the Spi phenotype (p. 83).
6.3.4 Cloning experiments with * insertion or replacement
vectors
A cloning experiment with a e vector can proceed along the same lines as with a plasmid
vector—the e molecules are restricted, new DNA is added, the mixture is ligated,
and the resulting molecules used to transfect a competent E. coli host (Figure 6.14a).
This type of experiment requires that the vector be in its circular form, with the cos
sites hydrogen bonded to each other.
Although satisfactory for many purposes, a procedure based on transfection is not
particularly efficient. A greater number of recombinants will be obtained if one or two
refinements are introduced. The first is to use the linear form of the vector. When the
linear form of the vector is digested with the relevant restriction endonuclease, the
left and right arms are released as separate fragments. A recombinant molecule can
100 Part I The Basic Principles of Gene Cloning and DNA Analysis
λ arms
EcoRI EcoRI
EcoRI
EcoRI
New DNA
cos cos
cos
cos cos
Catenane cos
EcoRI EcoRI
EcoRI
EcoRI
EcoRI, ligate
(a) Cloning with circular λ DNA
(b) Cloning with linear λ DNA
λ insertion factor
– circular form
cos cos
Transfect
E. coli
Infect E. coli
in vitro
packaging mix
Recombinant
molecule
Recombinant λ
New DNA
Ligate
Figure 6.14
Different strategies for cloning with a λ vector. (a) Using the circular form of λ as a plasmid. (b) Using left and right arms
of the λ genome, plus in vitro packaging, to achieve a greater number of recombinant plaques.
be constructed by mixing together the DNA to be cloned with the vector arms
(Figure 6.14b). Ligation results in several molecular arrangements, including catenanes
comprising left arm–DNA–right arm repeated many times (Figure 6.14b). If the inserted
DNA is the correct size, then the cos sites that separate these structures will be the right
distance apart for in vitro packaging (p. 81). Recombinant phage are therefore
produced in the test tube and can be used to infect an E. coli culture. This strategy, in particular
the use of in vitro packaging, results in a large number of recombinant plaques.
6.3.5 Long DNA fragments can be cloned using a cosmid
The final and most sophisticated type of e-based vector is the cosmid. Cosmids are
hybrids between a phage DNA molecule and a bacterial plasmid, and their design
centers on the fact that the enzymes that package the e DNA molecule into the phage
protein coat need only the cos sites in order to function (p. 21). The in vitro packaging
reaction works not only with e genomes, but also with any molecule that carries cos sites
separated by 37–52 kb of DNA.
A cosmid is basically a plasmid that carries a cos site (Figure 6.15a). It also needs a
selectable marker, such as the ampicillin resistance gene, and a plasmid origin of replication,
as cosmids lack all the e genes and so do not produce plaques. Instead colonies are
formed on selective media, just as with a plasmid vector.
Chapter 6 Cloning Vectors for E. coli 101
BamHI
ori
cos
BamHI
cos
Circular
pJB8
Restrict with
BamHI
BamHI BamHI
cos
cos cos
Linear pJB8 BamHI
BamHI BamHI BamHI
BamHI
New DNA
New
DNA
Ligate
In vitro package
Recombinant
cosmid DNA
Colonies containing circular
recombinant pJB8 molecules
Ampicillin medium
Infect E. coli
(a) A typical cosmid
(b) Cloning with pJB8
pJB8
5.4 kb
λ DNA
Catenane
λ particles
ampR
ampR
ampR
ampR
Figure 6.15
A typical cosmid and the way it is used to clone long fragments of DNA.
A cloning experiment with a cosmid is carried out as follows (Figure 6.15b). The
cosmid is opened at its unique restriction site and new DNA fragments inserted. These
fragments are usually produced by partial digestion with a restriction endonuclease, as
total digestion almost invariably results in fragments that are too small to be cloned with
a cosmid. Ligation is carried out so that catenanes are formed. Providing the inserted
DNA is the right size, in vitro packaging cleaves the cos sites and places the recombinant
cosmids in mature phage particles. These e phage are then used to infect an E. coli
culture, though of course plaques are not formed. Instead, infected cells are plated onto
a selective medium and antibiotic-resistant colonies are grown. All colonies are recombinants,
as non-recombinant linear cosmids are too small to be packaged into e heads.
6.4 8 and other high-capacity vectors enable
genomic libraries to be constructed
The main use of all e-based vectors is to clone DNA fragments that are too long to be
handled by plasmid or M13 vectors. A replacement vector, such as eEMBL4, can carry
up to 20 kb of new DNA, and some cosmids can manage fragments up to 40 kb. This
102 Part I The Basic Principles of Gene Cloning and DNA Analysis
Table 6.1
Number of clones needed for genomic libraries of a variety of organisms.
NUMBER OF CLONES*
SPECIES GENOME SIZE (bp) 17 kb FRAGMENTS† 35 kb FRAGMENTS‡
E. coli 4.6 × 106 820 410
Saccharomyces cerevisiae 1.8 × 107 3225 1500
Drosophila melanogaster 1.2 × 108 21,500 10,000
Rice 5.7 × 108 100,000 49,000
Human 3.2 × 109 564,000 274,000
Frog 2.3 × 1010 4,053,000 1,969,000
*Calculated for a probability ( p) of 95% that any particular gene will be present in the library.
†Fragments suitable for a replacement vector such as yEMBL4.
‡Fragments suitable for a cosmid.
compares with a maximum insert size of about 8 kb for most plasmids and less than 3 kb
for M13 vectors.
The ability to clone such long DNA fragments means that genomic libraries can be
generated. A genomic library is a set of recombinant clones that contains all of the DNA
present in an individual organism. An E. coli genomic library, for example, contains all
the E. coli genes, so any desired gene can be withdrawn from the library and studied.
Genomic libraries can be retained for many years, and propagated so that copies can be
sent from one research group to another.
The big question is how many clones are needed for a genomic library? The answer
can be calculated with the formula:
N =
where N is the number of clones that are required, p is probability that any given gene
will be present, a is the average size of the DNA fragments inserted into the vector, and
b is the total size of the genome.
Table 6.1 shows the number of clones needed for genomic libraries of a variety of
organisms, constructed using a e replacement vector or a cosmid. For humans and other
mammals, several hundred thousand clones are required. It is by no means impossible
to obtain several hundred thousand clones, and the methods used to identify a clone
carrying a desired gene (Chapter 8) can be adapted to handle such large numbers, so
genomic libraries of these sizes are by no means unreasonable. However, ways of reducing
the number of clones needed for mammalian genomic libraries are continually being
sought.
One solution is to develop new cloning vectors able to handle longer DNA inserts.
The most popular of these vectors are bacterial artificial chromosomes (BACs), which
are based on the F plasmid (p. 16). The F plasmid is relatively large and vectors derived
from it have a higher capacity than normal plasmid vectors. BACs can handle DNA
inserts up to 300 kb in size, reducing the size of the human genomic library to just
30,000 clones. Other high-capacity vectors have been constructed from bacteriophage
P1, which has the advantage over e of being able to squeeze 110 kb of DNA into its
ln(1 − p)
ln
A
1 − aD
C bF
Chapter 6 Cloning Vectors for E. coli 103
FURTHER READING
Further reading
Bolivar, F., Rodriguez, R.L., Green, P.J. et al. (1977) Construction and characterization of
new cloning vectors. II. A multipurpose cloning system. Gene, 2, 95–113. [pBR322.]
Frischauf, A.-M., Lehrach, H., Poustka, A. & Murray, N. (1983) Lambda replacement
vectors carrying polylinker sequences. Journal of Molecular Biology, 170, 827–842.
[The lEMBL vectors.]
Iouannou, P.A., Amemiya, C.T., Garnes, J. et al. (1994) P1-derived vector for the propagation
of large human DNA fragments. Nature Genetics, 6, 84–89.
Melton, D.A., Krieg, P.A., Rebagliati, M.R., Maniatis, T., Zinn, K. & Green, M.R. (1984)
Efficient in vitro synthesis of biologically active RNA and RNA hybridization probes from
plasmids containing a bacteriophage SP6 promoter. Nucleic Acids Research, 12,
7035–7056. [RNA synthesis from DNA cloned in a plasmid such as pGEM3Z.]
Sanger, F., Coulson, A.R., Barrell, B.G. et al. (1980) Cloning in single-stranded bacteriophage
as an aid to rapid DNA sequencing. Journal of Molecular Biology, 143, 161–178.
[M13 vectors.]
Shiyuza, H., Birren, B., Kim, U.J. et al. (1992) Cloning and stable maintenance of 300
kilobase-pair fragments of human DNA in Escherichia coli using an F-factor-based
vector. Proceedings of the National Academy of Sciences of the USA, 89, 8794–8797.
[The first description of a BAC.]
Sternberg, N. (1992) Cloning high molecular weight DNA fragments by the bacteriophage
P1 system. Trends in Genetics, 8, 11–16.
Yanisch-Perron, C., Vieira, J. & Messing, J. (1985) Improved M13 phage cloning vectors
and host strains: nucleotide sequences of the M13mp18 and pUC19 vectors. Gene,
33, 103–119.
capsid structure. Cosmid-type vectors based on P1 have been designed and used to clone
DNA fragments ranging in size from 75 to 100 kb. Vectors that combine the features
of P1 vectors and BACs, called P1-derived artificial chromosomes (PACs), also have a
capacity of up to 300 kb.
6.5 Vectors for other bacteria
Cloning vectors have also been developed for several other species of bacteria, including
Streptomyces, Bacillus, and Pseudomonas. Some of these vectors are based on
plasmids specific to the host organism, and some on broad host range plasmids able to
replicate in a variety of bacterial hosts. A few are derived from bacteriophages specific
to these organisms. Antibiotic resistance genes are generally used as the selectable
markers. Most of these vectors are very similar to E. coli vectors in terms of their general
purposes and uses.
Le phage Lambda était un choix idéal pour un vecteur de phage, car il était bien compris et facile à travailler. Plus important encore, le phage contient une région non essentielle interne vaste flanquée par des sites de clivage Eco RI. Par conséquent, cette région non essentielle peut être enlevée et l’ADN étranger inséré. Avant de l’ADN lambda coupé par Eco RI peut être utilisé pour le clonage, il était nécessaire d’éliminer les sites de clivage supplémentaires qui sont situés à dans les régions essentielles du génome lambda. Tout d’abord, un phage lambda hybride a été construit par recombinaison génétique in vivo. Cela manquait trois sites de clivage EcoRI à 0,438, 0,538 et 0,654 dans la région non essentielle mais a conservé les deux sites dans les régions essentielles. Ensuite, les deux autres les sites de clivage Eco RI ont été éliminés par mutation et sélection. Dans la sélection du phage ont été cyclée entre hôtes manquant et contenant les systèmes de modification de restriction EcoRI. Tous les phages avec le clivage des sites mutés de manière à être nonrecognizable par le système Eco RI aurait une plus grande probabilité d’échapper aux enzymes de restriction sur la croissance dans le second hôte. Après 10 à 20 cycles de cette sélection régime, Davis a trouvé un phage qui avait perdu l’un des deux sites R1, et après un 9 à 10 cycles supplémentaires, il a trouvé un mutant qui avait perdu la autre site. Ce phage mutant a ensuite été transformé en un vecteur de clonage utile par recombinaison avec le type sauvage lambda pour restaurer les trois EcoRI des sites de clivage situé dans la partie centrale du génome. L’ADN isolé des particules de phage lambda est linéaire et il peut être clivé par EcoRI pour donner les fragments centraux plus petits et les plus grands des bras gauche et droit. Les fragments d’ADN clonés pour être clivé par EcoRI peut ensuite être hybridé ensemble et ligaturé à droite purifié et bras gauche. Ce Soit l’ADN peut être utilisé tel quel pour transfecter des cellules rendues compétentes pour son prise ou il peut être encapsidé in vitro dans des têtes de phage et utilisé pour infecter
A J 2
Essentiel
b cI rouge int nin QSR
Essentiel
0,438 0,538 0,654 0,808 0,927
278 génie génétique et l’ADN recombinant
cellules. L’efficacité, par molécule de l’ADN, de l’emballage et l’infection est
beaucoup plus élevé que la transfection avec de l’ADN nu. Par conséquent, l’emballage est
utilisé lorsque le fragment à cloner est présent en quelques exemplaires seulement.
Vecteurs pour cellules supérieures
Le clonage de l’ADN dans des cellules plus élevées pose les mêmes problèmes que le clonage dans
bactéries. Les vecteurs doivent permettre simplement la purification de quantités considérables
de l’ADN, doit permettre la sélection des cellules transformées, et doit posséder
l’espace pour l’ADN inséré. Les vecteurs navettes, qui ont été largement
utilisé pour le clonage dans la levure, sont une solution élégante à ces exigences. Dans
plus de contenir les éléments de clonage de plasmide bactérien normal,
ils contiennent un réplicon de levure et un marqueur génétique sélectionnable dans la levure
(Fig. 9.10). En conséquence, de grandes quantités des vecteurs peuvent être obtenus
par croissance dans E. coli et ensuite transformée en levure. La capacité à
navette entre les bactéries et les levures permet d’économiser beaucoup de temps et de dépenses en
expériences de génie génétique.
Deux types d’origines de réplication de levure peuvent être utilisés dans la navette de levure
vecteurs. La première est une origine de réplication de l’ADN chromosomique de levure, également connue
comme un élément ARS. L’autre est l’origine 2 μ à partir des cercles. Ces
sont des éléments plasmidiques-like avec fonction inconnue qui se trouvent dans
Levure. Ils sont un peu plus stables que les vecteurs ARS. Nutritional
des marqueurs tels que l’uracile, l’histidine, la leucine et la biosynthèse du tryptophane
ont été utilisés comme gènes de sélection appropriée dans le auxotrophe
Levure.
Les virus constituent une base pour de nombreux vecteurs utiles dans plante supérieure et
des cellules animales. Par exemple, l’un des vecteurs les plus simples pour les mammifères
des cellules est le SV4O du virus simien. Il permet un grand nombre des mêmes clonage
que les opérations du phage lambda.
La terminologie utilisée avec des cellules de mammifères peut être déroutant.
« Transformation » peut signifier que les cellules ont reçu un plasmide. Ça peut
signifient également que les cellules ont perdu leur inhibition de contact. Dans cet état,
ils continuent de croître au-delà du stade de la monocouche de cellules confluentes à laquelle
ADN de levure
l’origine de replication
ampicilline
la résistance
tétracycline
la résistance
Origine de
Réplication de l’ADN
YRP17
TRP
E. coli
URA
ADN de levure
ADN de levure
Figure 9.10 La structure de
un vecteur pour la navette entre
E. coli et la levure. Il contient
les gènes qui permettent la réplication de l’ADN
et la sélection à la fois
organismes.
Vecteurs pour cellules supérieures 279
des cellules normales de mammifères cessent croissance. Transformation de la désinhibée
état de croissance peut résulter d’une infection par un virus de la tumeur comme causant
SV4O ou elle peut être le résultat d’une mutation du génome. Bien que la perte de
l’inhibition de contact pourrait être utile pour identifier les cellules qui ont incorporé
les hybrides SV4O ADN ou SV4O, cette propriété est d’une utilité limitée.
D’autres marqueurs génétiques sélectionnables convenables pour des cellules de mammifères sont
Obligatoire.
Un gène utile pour les sélections dans les cellules de mammifères a été le
le gène de la thymidine kinase, car les cellules TK + peuvent être sélectionnées en cultivant
elles dans un milieu contenant de l’hypoxanthine, de l’aminoptérine et de la thymidine.
A l’inverse, les cellules TK- peuvent être sélectionnées en les cultivant dans un milieu
contenant de la bromodésoxyuridine (Fig. 9.11). En outre, les virologues
ont découvert précédemment que les codes de virus de l’herpès simplex de son
propre thymidine kinase. Par conséquent, le génome viral peut être utilisé en tant que
source concentrée du gène sous une forme exprimable pour le clonage initial
expériences.
Bien que le gène de la thymidine kinase a été utile pour la sélection de cellules
qui ont absorbé l’ADN étranger, un gène de sélection qui ne nécessite pas
l’isolement préalable d’une thymidine kinase mutant négatif dans chaque cellule
ligne serait également utile. E. coli enzyme xanthine-guanine
Hypoxanthine + Aminopterin + thymidine
Précurseurs IMP XMP GMP
xanthine
(Voie faible
les cellules de mammifères)
xanthine phosphoribosyl
transférase
GTP
AMP
thymidine
kinase
Aminopterin
dUMP dTMP (BrdU-MP, toxiques)
mycophénolique
acide
Thymidine (BrdU)
hypoxanthine
Brdu TK
TK +
–
Sélectionner
Figure 9.11 Les voies métaboliques impliquées avec quelques gènes sélectionnables dans
les cellules de mammifères. IMP-inosine monophosphate, monophosphate XMP-xanthine,
GMP-guanosine monophosphate, duMP-désoxyuridine monophosphate,
dTMP-désoxythymidine monophosphate, Brdu-bromodésoxyuridine, Brdu-MP,
monophosphate bromodésoxyuridine. blocs aminoptérine tétrahydrofolate réductase,
qui est nécessaire pour la synthèse de l’IMP et dTMP et mycophénolique
l’acide bloque la synthèse de XMP.
280 génie génétique et l’ADN recombinant
gène transférase phosphoribosyl semble répondre à ces exigences.
Le produit protéique des fonctions de gènes dans des cellules de mammifères et
permet la croissance sélective des cellules non mutant qui contiennent l’enzyme
(Fig. 9.11). Le milieu de croissance requis contient la xanthine, l’hypoxanthine,
aminoptérine, et de l’acide mycophénolique. D’autres gènes dominants
utile pour la sélection des cellules transformées sont des mutants de dihydrofolate
réductase qui est résistante au methotrexate, un inhibiteur puissant de la
Enzyme de type sauvage, et la kanamycine-néomycine phosphotransférase. le
celle-ci est une enzyme dérivée d’un transposon bactérien et confère
résistance aux bactéries, des levures, des plantes et des cellules de mammifères à un composé
appelé G418. Bien entendu, pour une expression appropriée dans les cellules supérieures, la
gène doit être relié à une unité de transcription appropriée et doivent
contenir les signaux d’initiation et de polyadénylation traduction requis.
mettre l’ADN
Chapitre 1: la technologie de l’ADN recombinant
Définition :
Les termes du génie génétique et l’ADN recombinant se réfèrent à des techniques dans lesquelles l’ADN peut être coupé, rejoint, sa séquence déterminée ou la séquence d’un segment modifié en fonction de l’utilisation prévue. Par exemple, un fragment d’ADN peut être isolé d’un organisme, joint à d’autres fragments d’ADN, et mis dans une bactérie ou d’autres organismes. Dans un autre exemple du génie génétique un fragment d’ADN, souvent un gène entier, peut être isolé et sa séquence nucléotidique déterminée ou sa séquence nucléotidique peut être altérée par des méthodes mutagènes in vitro. Ces activités connexes en génie génétique ont deux objectifs fondamentaux :
1) pour en apprendre davantage sur les œuvres de la nature.
2) trouver les moyens de faire usage de ces connaissances à des fins pratiques.
L’isolement du phage spécialisé de transduction qui portait les gènes de l’opéron lac a été une avancée particulièrement importante, car ces phages ont fourni un enrichissement de plus de 100 fois des gènes lac par rapport à l’ADN chromosomique. Il a également stimulé une grande variété d’études importantes qui ont favorisé grandement notre compréhension de la régulation des gènes ainsi que favorisé le développement de nombreuses techniques de génie génétique importantes. Maintenant le génie génétique permet les mêmes sortes d’études à effectuer sur un gène à partir de pratiquement tout organisme.
La deuxième raison majeure de l’intérêt du génie génétique est la synthèse économique de protéines, difficiles ou impossibles à purifier à partir de leurs sources naturelles. Ces protéines peuvent être des antigènes pour une utilisation dans l’immunisation, les enzymes pour une utilisation dans les procédés chimiques ou des protéines spécialisées à des fins thérapeutiques. Les séquences d’ADN cloné peuvent également être utilisées pour la détection des défauts chromosomiques et dans des études génétiques. Beaucoup de recherches en génie génétique ont également été réalisées dans les usines avec l’espoir d’améliorer les méthodes génétiques traditionnelles de modification des cultures. Un second objectif est l’introduction de la résistance aux herbicides dans les cultures souhaitées. Cela permettrait la pulvérisation contre les mauvaises herbes dans les champs même pendant la croissance au lieu de le faire avant la plantation. Le génie génétique de l’ADN implique généralement les étapes suivantes :
1) L’ADN pour l’étude doit être isolé et libéré des contaminants. Il doit être possible de couper cet ADN à des endroits spécifiques afin de produire des fragments contenant des gènes ou parties de gènes.
2) Ensuite il doit être possible de rejoindre les fragments d’ADN pour former des molécules d’ADN hybrides.
3) Les vecteurs doivent exister pour que les fragments assemblés puissent être introduits dans des cellules par le procédé appelé transformation.
4) Les vecteurs doivent avoir deux propriétés. Tout d’abord ils doivent fournir l’ADN autonome du vecteur pour la réplication dans les cellules et, d’autre part, ils doivent permettre une croissance sélective des seules cellules qui ont reçu les vecteurs.
Ce chapitre décrit les étapes fondamentales du génie génétique ainsi que la technique essentielle de la détermination de la séquence nucléotidique d’un fragment d’ADN
L’isolement de l’ADN :
L’ADN cellulaire, chromosomique ou non chromosomique, est le point de départ de nombreux expériences de génie génétique. Un tel ADN peut être extrait et purifié par les techniques classiques de chauffage des extraits cellulaires en présence de détergents et après élimination des protéines par extraction au phénol. Si des polysaccharides ou des ARN contaminent l’échantillon ils peuvent être éliminés par centrifugation à gradient de densité d’équilibre au chlorure de césium. Deux types de vecteurs sont communément utilisés : des plasmides et des phages. Un plasmide est un élément d’ADN semblable à un épisome qui se reproduit de manière indépendante du chromosome. Habituellement les plasmides sont de petites tailles, 3000 à 25.000 paires de bases et circulaire.
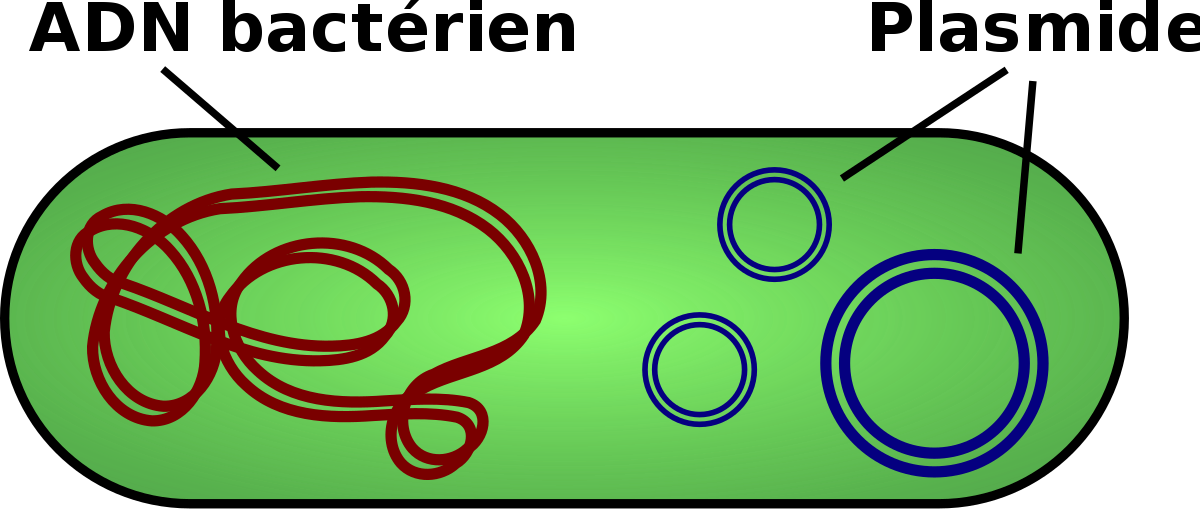
En général, le phage lambda ou des dérivés apparentés sont utilisés comme vecteur pour des Escherichia coli, mais pour clonage dans d’autres bactéries, comme Bacillus subtilis, d’autres phages sont utilisés. Dans certains cas un plasmide peut être développé qui se réplique de façon autonome en dehors de plus d’un organisme hôte. Ces vecteurs « navettes » sont importants dans l’étude des gènes des eucaryotes, nous allons les examiner plus tard. Le plus souvent la purification complète de l’ADN de plasmide est inutile et l’ADN utilisable peut être obtenu simplement par la lyse des cellules en éliminant partiellement l’ADN chromosomique et la plupart des protéines. Les constructions complexes d’ADN nécessitent souvent l’ADN très pur pour éviter l’interférence des nucléases exogènes ou l’inhibition des enzymes sensibles.
L’isolement de l’ADN plasmidique :
La purification complète de l’ADN plasmidique nécessite généralement plusieurs étapes. Après que les cellules sont ouvertes avec le lysozyme qui digère la paroi cellulaire et que les détergents sont ajoutés pour solubiliser les membranes et inactiver certaines protéines, la plupart des ADN chromosomiques sont éliminés par centrifugation. Pour de nombreux objectifs des méthodes chromatographiques peuvent être utilisées pour effectuer la purification. Lorsque la pureté la plus élevée est nécessaire le plasmide est purifié par la densité d’équilibre de centrifugation à gradient. Cela se fait en présence de bromure d’éthidium. Tout ADN chromosomique restant avec le plasmide aurait été fragmenté et sera linéaire, alors que la plupart des ADN plasmidique seront circulaires car liés de manière covalente. Comme nous l’avons vu précédemment l’intercalation de bromure d’éthidium détord l’ADN.
Pour une molécule circulaire cette détorsion génère sur-enroulement alors que pour une molécule linéaire la détorsion n’a pas d’effets majeurs. Par conséquent un ADN linéaire peut intercaler plus du bromure d’éthidium qu’une molécule circulaire. Etant donné que le bromure d’éthidium est moins dense que l’ADN, les molécules d’ADN linéaires intercalées avec bromure d’éthidium « flottent » par rapport à l’ADN circulaire et donc les deux espèces peuvent facilement être séparées. Suivant la centrifugation, les deux bandes d’ADN sont observées par les UV éclairant la lumière du tube. La fluorescence naturelle du bromure d’éthidium est augmentée de 50 fois par intercalation dans l’ADN et le Phage lambda peut également être partiellement purifié par des techniques rapides et éliminer les débris cellulaires et la plupart des contaminants. Une purification plus complète peut être obtenue en utilisant leur densité unique de 1,5 g/cm3, qui est à mi-chemin entre la masse volumique de protéines, de 1,3 et la densité de l’ADN, 1.7. Le phage peut être isolé par la centrifugation à gradient de densité d’équilibre dans laquelle la densité à mi-chemin entre le haut et le bas du tube de centrifugation est de 1,5. Eux aussi peuvent être facilement observés dans la centrifugeuse. Ils forment une bande bleue qui résulte de la dispersion préférentielle des longueurs d’onde plus courtes de la lumière connue sous le nom d’effet Tyndall. Ce même phénomène est la raison pour laquelle le ciel est bleu et les couchers de soleil sont rouges.
La biologie des enzymes de restriction :
Dans cette section, nous allons d’abord voir la biologie des enzymes de restriction et puis revenir à leur utilisation pour couper l’ADN. On a maintenant trouvé un grand nombre d’enzymes qui coupent l’ADN à des endroits spécifiques. Pour la plupart ces enzymes proviennent de bactéries. Elles sont appelées des enzymes de restriction parce que, dans les quelques cas qui ont été soigneusement étudiés, le clivage enzymatique de l’ADN fait partie du système de restriction-modification de la cellule. Le phénomène de restriction-modification des bactéries est un système immunitaire à une petite échelle pour une protection contre l’infection par l’ADN étranger. Contrairement à des organismes supérieurs dans lesquels l’identification et l’inactivation de l’invasion des parasites, des bactéries ou des virus peuvent être effectuées de manière extracellulaire, les bactéries peuvent se protéger après que l’ADN étranger est entré dans leur cytoplasme. Pour cette protection de nombreuses bactéries marquent spécifiquement leur propre ADN par la méthylation des bases sur des séquences particulières avec des enzymes de modification. L’ADN qui est reconnu comme étranger par l’absence de groupes méthyle sur ces mêmes séquences est clivé par les enzymes de restriction puis dégradé par les exonucléases aux nucléotides. Moins d’un phage sur 10000 sont erronément méthylés et sont capables de croître et de lyser un E.coli protégé par certains systèmes de modification-restriction. En outre les bactéries se protègent de l’ADN végétal et animal. L’ADN de beaucoup d’ animaux et de végétaux est méthylé sur la cytosine dans les séquences CpG. Beaucoup de souches de bactéries contiennent également des enzymes qui clivent l’ADN lorsqu’il est méthylé sur des positions spécifiques.
Arber a étudié la restriction du phage lambda dans E. coli et a constaté que La souche E. coli C ne contenait pas de système de modification-restriction. Souche B a un système de modification-restriction, et pourtant un autre reconnaît et méthyle une séquence nucléotidique différente dans la souche K-12. Phage P1 possède un système de modification- restriction propre à elle même qui est superposable au système de restriction-modification de l’hôte dans lequel elle est un lysogène.
Soit la notation λ-C représente la croissance de phage lambda qui a été cultivé sur La souche E. coli C. L’infection des souches B, K-12, et C avec λ sur diverses souches va se faire à différentes vitesses :
La possession d’un système de modification-restriction introduit des complexités dans le processus de replication de l’ADN. Imaginez que le double brin d’ADN contient des groupes méthyle sur les deux brins de l’ADN au niveau d’une séquence de reconnaissance. la réplication de l’ADN crée un duplex dans lequel l’un des brins des duplexes fille dans un premier temps n’a pas été modifié .
Cet ADN demi-méthylé ne doit pas être reconnu comme un ADN étranger et clivé, mais doit être reconnu comme «soi» et méthylé.
Par conséquent, le système de restriction- modification fonctionne comme un micro-ordinateur, reconnaissant les trois états différents de la séquence de reconnaissance il réagit de trois manières différentes :
1) Si la séquence n’ est pas méthylé, les enzymes la clivent.
2) Si l’ADN est méthylé sur l’un des deux brins, le système de modification méthyl l’autre brin.
3) si l’ADN est méthylé sur les deux brins, les enzymes ne font rien.
Une séquence de reconnaissance palindromique simplifie le fonctionnement du système de modification-restriction. Un palindrome est une séquence qui se lit de la meme manière de gauche à droite et de droite à gauche, comme les mots « REPAPER et RADAR ». Comme les brins d’ADN possèdent une direction, on considère une séquence d’ADN à palindrome si elle est identique à la lecture de 5 ‘à 3’ sur le brin supérieur et le brin inférieur . les Palindromes, peuvent être de toutes tailles, mais la plupart de ceux qui sont utilisés en tant que séquences de reconnaissance de modification-restriction sont quatre, cinq, six, et rarement, huit bases. En vertu des propriétés de palindromes, les deux filles de palindromes répliquées ont des séquences identiques, et donc l’enzyme de modification doit reconnaître et méthyler un seul type de substrat (Fig. 9.4).
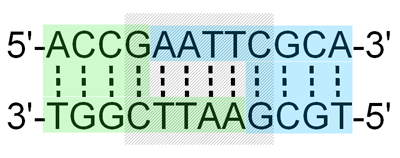
Comme nous l’avons déjà vu précédemment pour la reconnaissance des séquences non palindromic, il faudrait que l’enzyme de modification reconnaîsse à la fois les deux séquences différentes. On peut supposer que les protéines dimères sont utilisés pour les reconnaître. Les enzymes de restriction sont divisés en trois catégories principales. les enzymes
1) Classe I : ces enzymes sont formées de 3 sous-unités (d’une sous-unité de clivage,une sous-unité de méthylation et une sous-unité de reconnaissance des séquences). Ces enzymes clivent à
des sites éloignés de leurs séquences de reconnaissance et ne seront pas discuté ici même si elles ont été les premiers à etre découvertes.
2) Classe II :les enzymes de classe II possèdent leur sous unité de reconnaissance des séquences et leur sous unités de clivage ensemble. Ils coupent près de leur séquence de reconnaissance et sont le plus utilisé dans le génie génétique.
3) classe III possèdent 3 sous-unités (la sous-unité de clivage associée à celle de reconnaissance et la sous-unité de méthylation). Ces enzymes coupent près de leur site de reconnaissance.
Une enzyme de restriction dans une cellule est une bombe à retardement parce que les principes physicà-chimiques
limitent leur spécificité pour la liaison. Si une enzyme de restriction se liait à une séquence erronée, et comme une bactérie typique contient environ 4 × 1000000 de telles séquences, très probablement la séquence ne serait pas méthylé et l’enzyme pourrait cliver l’ADN et la cellule mourrerait. l’observation, cependant, est que les cellules contenant des enzymes de restriction ne meurent pas plus vite que les cellules sans enzymes de restriction. Comment, alors, est généré la spécificité extraordinairement grande des enzymes de restriction ?
La coupure d’ADN avec des enzymes de restriction :
Les enzymes de restriction fournissent un outil nécessaire pour couper des fragments de l’ADN à partir de molécules plus grandes. Leur spécificité permet une très grande sélectivité, et leur grand nombre ( plus d’une centaine de restrictions différentes), permet une trés grande variétés de choix des sites de clivage utilisés. Souvent, les fragments peuvent être produits avec des points d’extrémité situés à moins de 20 paires de bases de n’importe quel emplacement souhaité.
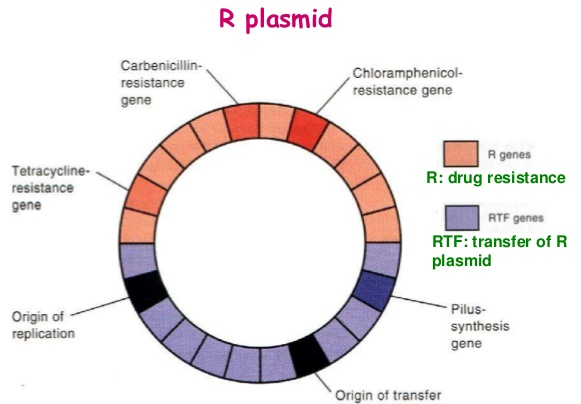
Une des propriétés les plus utiles des enzymes de restriction pour le génie génétique se trouve dans le système de modification-restriction produit par Plasmide R d’E.coli. L’enzyme de restriction correspondante est appelée Eco RI. Au lieu de cliver au centre de sa séquence de reconnaissance palindromique, cette enzyme coupe à l’extrémité et produit des extrémités à 4 bases complémentaires.Ces extrémités « collantes » sont les plus utiles dans la technologie de l’ADN recombinant. Elles peuvent être re-soudés à basse température comme les extrémités « collantes » du phage lambda. Cela permet l’assemblage efficace des fragments de l’ADN au cours des étapes de soudures . Environ la moitié des enzymes de restriction, actuellement connu peuvent générer des extrémités en surplomb ou collantes. Dans certaines situations, un fragment d’ADN peut même être agencé de telle manière à avoir deux types de collant différents.
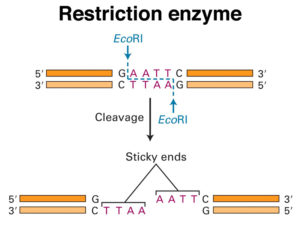
L’isolement des fragments d’ADN :
Après le clivage de l’ADN par des enzymes de restriction ou d’autres manipulations (cf plus tard) les fragments d’ADN doivent souvent être isolés. Heureusement, le fractionnement en fonction de la taille est particulièrement facile parce que, comme indiqué précédemment, l’ADN possède un rapport charge sur masse constant et les fragments d’ADN double brin de la même longueur ont la même forme et donc migrent pendant l’électrophorèse à une vitesse pratiquement indépendant de leur séquence. En général, plus l’ADN est grand, plus sa vitesse de migration est lente. Une très grande résolution peut être obtenue en électrophorèse par example deux fragments dont les tailles diffèrent de 0,5% peuvent être séparés si elles se trouvent dans une plage de 2 à 50 000 paires de bases. Une plage typique pour une séparation de taille adéquate pourrait être de 5 à 200 paires de bases ou 50 à 1000 paires de bases, et ainsi de suite. Le matériau à travers lequel l’ADN est soumis à une électrophorèse doit posséder des propriétés particulières. Il doit être peu coûteux, facilement utilisable, non chargée, et il devrait former un réseau poreux. Deux matériaux répondent aux exigences: agarose et polyacrylamide. Après l’électrophorèse, les bandes formées par les différentes tailles des fragments peuvent être localisés par autoradiographie si l’ADN avait été radiomarqué avant la séparation. Habituellement 32PO4 est une étiquette commode parce que le phosphate se trouve dans l’ARN et l’ADN, 32P émet notamment des électrons énergétiques qui les rend facilement détectables, et enfin 32P a une demi-vie courte de sorte que la plupart des atomes radioactifs dans un échantillon désintégreront dans un délai raisonnable. L’isotope 33P est également utilisé. Sa désintégration bêta est plus faible, et il a une demi-vie de 90 jours. Souvent, l’ADN est suffisamment présent et il peut être directement détecté par coloration avec éthidium bromure. La fluorescence accrue du bromure d’éthidium intercalées dans l’ADN par rapport à sa fluorescence dans la solution permet la détection d’aussi peu que 5 ng d’ADN dans un groupe. Après la séparation par l’ électrophorèse et la détection de l’ADN, les fragments souhaités peuvent être isolé à partir du gel.
Souder les fragments d’ADN :
Après la coupure et la purification de l’ADN nous allons discuter de l’assemblage des molécules d’ADN. In vivo, l’enzyme ADN ligase répare les entailles survenant sur la squelette de l’ADN. Cette activité peut également être utilisés in vitro pour la jonction de deux molécules d’ADN. Deux exigences doivent être respectées. D’abord, les molécules doivent être les substrats corrects, autrement dit, ils doivent posséder des groupes hydroxyle 3′- et 5′-phosphate. Ensuite les groupes sur les molécules à assembler doivent être correctement positionnés par rapport à l’autre. Le procédé pour produire le bon positionnement présente deux variantes: soit hybrider les fragments ensemble par leurs extrémités collantes complémentaires :
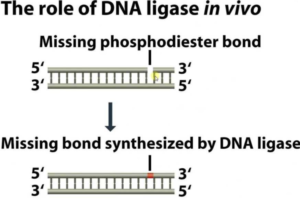
et, si l’assamblage concerne des fragments coupés avec bouts francs , d’utiliser des concentrations très élevées de fragments qui, de temps en temps,seront spontanément face à face avec positions correctes.De nombreuses enzymes de restriction telles que EcoRI produisent des extrémités collantes à quatre bases qui peuvent être soudées ensembles. Comme les extrémités collantes sont généralement seulement quatre paires de base, la baisse de la température à environ 12 ° C, facilite le processus d’hybridation-ligature. Les extrémités franches de molécules d’ADN qui sont générés par certaines enzymes de restriction génèrent des problèmes. Une solution consiste à convertir les bouts francs des molécules à des bouts collants par l’enzyme terminale transférase. Cette enzyme ajoute des nucléotides à l’extrémité 3′ de l’ADN. Une queue Poly-dG peut être ajouté à un fragment et une queue poly-dC peut être ajouté à l’autre et les fragments peuvent ensuite être hybridés ensemble
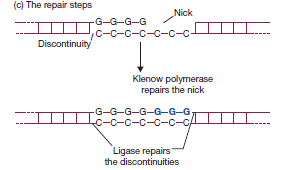
Si les queues sont assez longues, le complexe peut être introduit directement dans les cellules, où les lacunes et les pseudos seront remplis et scellés par le système enzymatique des cellules. Le plus souvent, la réaction en chaîne par polymerase ( PCR) serait utilisée pour générer des extrémités souhaitées sur les molécules. En outre les molécules ayant un bout franc peuvent également être soudées
par l’ADN ligase. Bien que cette méthode est très simple, elle souffre de deux inconvénients: Elle nécessite des concentrations élevées d’ADN et de l’ADN ligase pour que la réaction s’initie et meme dans ces conditions l’efficacité reste faible. En outre, il est difficile d’exciser le fragment du vecteur . Les Lieurs peuvent également être utilisés pour générer des molécules auto-complémentaires monocaténaires. Les Linkers sont des ADN courtes avec bouts francs , contenant la séquence de reconnaissance d’une enzyme de restriction qui produit des extrémités cohésives. La ligature des lieurs à des fragments d’ADN se produit avec un rendement assez haut car des concentrations molaires élevées des lieurs peuvent facilement être obtenues. Une fois que les lieurs ont été relié au segment d’ADN, le mélange est digéré avec l’enzyme de restriction qui coupe les lieurs et génère des extrémités collantes. Ainsi une molécule d’ADN à bout franc est converti en une molécule collante à composition non limitée qui peuvent facilement être jointes à d’autres molécules d’ADN.
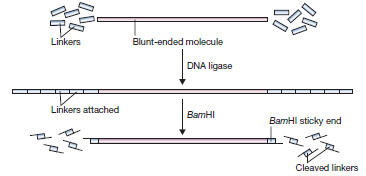
Chapter 5 : asymmetric synthesis – formation of optically-pure compounds
There are two ways to obtain such compounds. The first method is called the resolution in which the reactants we are starting with are either racemic or achiral. We separate the enantiomers after the production of both enantiomers. This method is vastly used in industries even if it doubles the volumes. They try to use the inactive enantiomer in another process. During a diastereoisomeric resolution, the separation of the enantiomers is made by the addition of an optically pure enantiomer called the resolution agent.
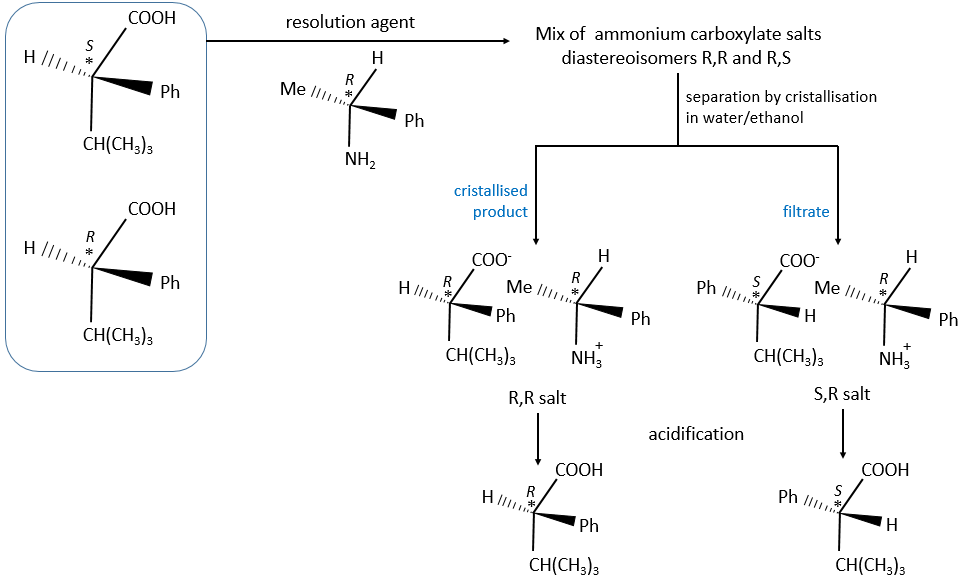
The formed salt is a racemic melange of diaseteoisomers that can now be separated.
The separation of the enantiomers can also be done via the kinetic splitting that is based on the difference of reaction speed of the enantiomer with a given chiral reactant.
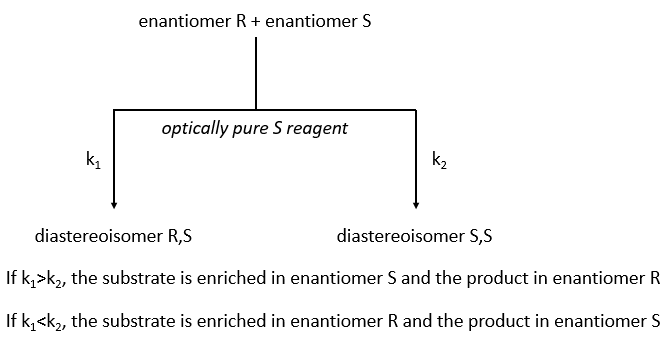
The speed of reaction has to be very different for the two enantiomers. If we put 0.5 equivalent of the chiral reactant, the enantiomer that reacts faster will from the diastereoisomer and the other enantiomer remains intact (ideal case). The conversion ratio is the proportion of the intact enantiomer over the proportion of the formerd diastereoisomer.
The second way is to directly produce the chiral species. This technique evolved over the years and began with the use of a chiral substrate. Around 1980 a chiral auxiliary was used. In this decade, they also used chiral reactants and after 1990 we started to use chiral catalysts. We will describe those techniques soon. First we will talk about the chiral pool.
The chiral pool
The chiral pool refers to natural compound that are optically active and can be produced at large scale (and thus are cheap). It can be aminoacids, peptides, metabolites, etc. We can use those compounds as starting reactants to obtain a chiral final product. The stereocentres already present on the natural compound will control the stereoselectivity of the reactions and lead to one single enantiomer. We talk about a hemisynthesis (because the starting reactant has not be chemically produced).
Amino-acids
At the exception of the glycine, the 20 amino-acids are chiral and available with a good ee (enantiomeric excess). They are bifunctional and levogyre but an enzymatic method can reverse their configuration.
Hydroxy-acids
They wear one or more OH and COOH and can have several stereocentres.
Carbohydrates
They are very cheap but we have to modify them and to protect some groups to use them correctly.
Terpenes
Most of them are used as precursors of agent of resolution or as ligands during asymmetric catalysis but they can also be used as starting reactant of other terpenes. They are cheap and available in large quantities. Their cyclic structure allows a better control of the strereoselectivity. However, they don’t have a lot of functional groups and their optic purity is not always very good.
Alcaloïdes
These secondary metabolites possess a nitrogen aromatic and are used as chiral catalysts or agents of resolution.
Chapter 4 : asymmetric synthesis – analytical methods of determination of the enantiomeric excess
A chiral agent is always necessary to differentiate the enantiomers. The most used methods to determine the ee are
- the GC with a chiral stationnary phase,
- HPLC with a chiral stationnary phase,
- NMR with chiral lanthanides.
Gazeous chromatography
The stationnary phase of the column is chiral and only composed of one enantiomer (for instance the R configuration). When a racemic melange passes through the column, the enantiomers R’ and S’ have a different interaction with the chiral stationnary phase with which they form diastereoisomeric complexes.
The main interactions can be H bonds, sterical interactions or dipole-dipole interactions. As the interactions have not the same strength, the enantiomers R’ and S’ have a different retention time and exit the column separately, giving two peaks.
The advantages of this method is that one the condition of experimentation are setted up, the analysis is fast and has a high sensibility. It can detect an enantiomer even if its proportion in the solution is about 0.5%. Moreover, the analysis separates the species composing the sample and we can select the portion of the solution that we want to analyse next.
When we use this method, the racemic melange has first to be tested to be sure that the enantiomers can be separated and that the ratio of the areas of the peaks is 50:50.
NMR
This technique is not as sensible as the GC but it is very fast and cheap.
Normally, enantiomers have the same NMR spectrum because the interatomic distances are identical for the two enantiomers. Diastereoisomers have spectra that can be distinguished. To distinguish two enaintiomers we will thus transform them into diastereoisomers.
![]()
To do so, we use two types of chiral reactants.
- chiral agent of derivatisation
- chiral lanthanides
A chiral agent of derivatisation is a chiral compound optically pure that interacts with the enantiomers. The most used one is the Mosher’s acid (α-methoxy-α-trifluoromethylphenylacetic acid (MTPA)).
This reactant forms a covalent liaison with the enantiomer to form diastereoisomers with an important difference of chemical displacement. The ratio between the areas gives the enantiomeric excess.
The method with chiral lanthanides is more used than the method with a chiral agent of derivation.
Lanthanides allow a widening of the NMR spectrum by the displacement of the signals towards the low fields. The salt of lanthanide is added bit by bit to the sample to observe the separation of the signals. They form a complex with the enantiomer without covalent liaison but still give an important separation of chemical displacement.
The disadvantages of this method is that it takes more time (the salt is added slowly) and that the salt is expensive.
Chapter 3 : asymmetric synthesis – stereoisomery
Editor’s note: It is a good exercise to determine the configuration (R or S) of each chiral centre presented in this course.
Some molecules have the same composition in terms of atoms but differ by their placement. Those molecules are called conformers (of configuration). Amongst them we distinguish enantiomers and diastereoisomers. The distinction was already explained in a previous section and we will explain it here too. Despite one slight difference of arrangement, the activity of two enantiomers can often differ signifficantly, especially in biology where the damages can be important. Pharmaceutical industries have to separate the enantiomers in the medecines or prove that the other enantiomer is truely inactive. Separation methods exist to isolate one of the enantiomer but it is one more step in the process and there is still 50% of product that is simply lost. For that reason, the field of asymmetric synthesis developped quickly in the current of the 1980’s.
Examples of problems due to the enantiomer
A melange of enantiomers can be less active for antagonism reasons: one enantiomer blocks the activity of the other enantiomer. For instance, the R pheromone of japanese cockroaches becomes totally inactive if the isomer S is present (the notion of R,S, (+) and (-) will be explained soon), even in a proportion of 1%. The S isomer can fix itself on the same receptor than the R isomer but it is inactive. Once it is on the receptor is doesn’t leave, making the R isomer inactive.
The use of a melange can be dramatic. Thalidomide is a medecine that treat the symptoms of sleeping troubles and of depression. The molecule is active even in presence of its isomer but the isomer causes damages to the phoetus of pregnant women. Disformed babies were born because the enantiomers were not separated at this time.
On the figures above, we use three types of lines to represent liaisons:
- a full line indicates either that the liaison is in the plane of the sheet or that its direction outside this plane has no interest,
- black triangles mean that the liaison goes out from the plane of the sheet (point side of the triangle) towards the reader (base of the triangle). It can be seen as a full line we see closer and closer,
- parallels lines in the shape of a triangle is the opposite of one black triangle: the liaison goes from the plane of the sheet (small side) to away from the reader (large side).
Another example is the penicillamine. One isomer is an antidote to poisoning by Pb, Au or Hg. The enantiomer can make you blind.
No need to say that it is better to avoid any melange of the two enantiomers. Eniantiomers can also show big difference in taste or smell because they will activate different receptors in our nose/on our tongue.
Reminder on the stereoisomery
Isomers are compounds of same composition but with a different structure. Isomers of constitution have not the same arrangement of atoms. A simple example is the n-butane and the isobutane.
Amongst the isomers, we distinguish stereoisomers. In stereoisomers, the sequence of atoms is identical but the spacial arrangement of the atoms is different. Stereoisomers of configuration can be separated and isolated. To pass from one stereoisomer of configuration to another one, one liaison has to be broken. For instance, the configuration around a C=C liaison can be Z or E, but we have to break the π liaison to go from one to the otherone.
Stereoisomers of conformation, or conformers, are different arrangements of the same molecule obtained by the rotation of one (or more) σ liaison. There is a barrier of energy for this rotation, which can be huge if the rotation is made difficult by the presence of a sterically hindred substituent. Moreover the rotation depends on the temperature. An simple example is the chair-boat conformers of the hexane.
The chair conformation is favoured by 6kCal/mol because all the substituents are in staggered position while they are all in eclipsed position in the boat conformer, leading to a bigger sterical hindrance. There is an equilibrium between the two conformers which is completely displaced towards the chair conformation at ambiant conformation. If we decrease the temperature we can observe both conformers and even separate them. The barrier of energy depends on the substituents of the hexane and the compound is more stable when big substituents are on an equatorial spot because the steric hindrance is smaller there. On the boat conformation, the steric hindrance is always more important because all positions are eclipsed.
Sometimes the rotation of one liaison is so much hindered that the life time of the isomer is long enough to be considered as a stereoisomer instead of as a conformer. As a general rule, we say that we are in presence of conformers if ∆G¹<22.6kCal/mol. If it is larger, then we are in presence of stereoisomers (atropoisomers: denial by steric hindrance). This value corresponds to a life-time of approximatively 1 hour at 25°C.
Binaftines are composed of two benzenes bound by one σ liaison. If there is no substituent on the aromatic cycles, the molecule is plane. The liaison between the cycles is not completely one σ liaison as all the π liaisons are conjugated. If there is one or more substituent on A, B, C or D, the steric hindrance doesn’t allow the planarity of the molecule and it becomes very difficult for the σ liaison to make a complete rotation. For instance, the binaftol cannot turn at ambiant temperature.
Chirality
The chirality is the property of an object not to be superimposable to its image in a mirror. The usual example of chiral objects is our hands. The left hand is the mirror image of our right hand but we cannot superimpose them. Indeed, if our palms are in front of us, the fingers are not at the same place. For instance thumbs are not at the same side of the hand. Even if we rotate one hand by 180°, fingers are at the same place but not in the same direction. The word itself, chirality, is derived from the greek word for hand. As a general rule, if a molecule has no plane or centre of symmetry, this molecule is chiral. Yet, there can be an axis of symmetry in achiral molecules.
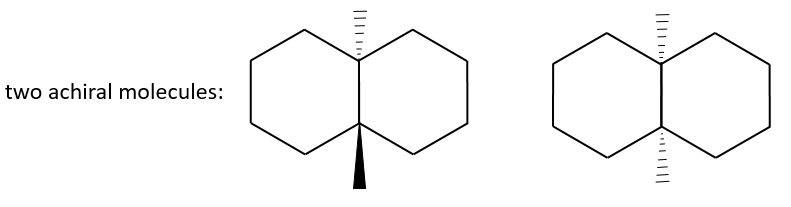
The stereogenic unity responsible for the chirality of the molecule can be
- an atom,
- an axis
- a plane
- the helicity of the molecule
There are thus 4 main types of chiral molecules.
Amongst the stereoisomers of configuration, we distinguish the enantiomers that are chiral and the diastereoisomers that are not chiral and not identical.
Enantiomery
The distances between the atoms are identical in enantiomers. As a result, the chemical and physical properties of enantiomers are identical as long as there is no external chiral influence. A particular property of chiral molecules is their optical activity or rotatory power [α]. A chiral substance crossed by a polarised light induces a rotation of the plane of polarisation of the light in one direction.
The direction in which the light is deflected depends on the enantiomer. For instance the 1,1-aminophenylethane has two enantiomers the optical activity of which is [α]D=39 and [α]D=-39 if illuminated by sodium at 20°C (the D indicates these experimental conditions). The specific activity of one enantiomer is given by
where λ is the wavelenght of the polarised light, T the temperature, α the observed angle of rotation, b the length of the cell containing the substance and c the concentration of the solution. The cell in itself has to have no optical activity.
If the value is negative, the compound is levogyre (noted l or (-)) and if the value is positive, the compound is dextrogyre (d or (+)). In the case of the 1,1-aminophenylethane, the R enantiomer is dextrogyre with an optical activity of [α]D=+39 and the S enantiomer is levogyre [α]D=-39). Unfortunatly, there is no correlation between the fact that the enantiomer is R/S and levogyre/dextrogyre.
If the solution is composed of a 50:50 racemic melange of two enantiomers, the light is not deviated and [α]=0. In fact the light is deviated at the miscroscopic scale but in average there is no deviation at the detector because the rotatory powers of the enantiomers counterbalance each other. We define the optical purity (also called the enantiomeric excess ee) as
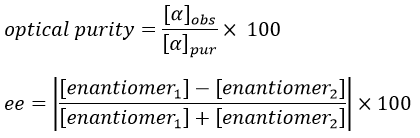
Stereogenic centres
1) centre of chirality
An asymmetric carbon (or any other tetravalent atom) on which there are 4 different substituents is a centre of chirality. The absolute configuration is the real disposition of the atoms around the atom of carbon. The carbon is S or R, what can be determined with the rule of Cahn, Ingold and Prelog (CIP)(there is a second method that can be used if you prefer it).
The first thing to do is to find the atomic number of the atoms bound to the assymetric carbon (consider zero for a lone pair). If two substituents have the same atomic number, we look at their substituents (starting with the highest atomic numbers), etc.
We can name them A, B, C and D starting by the highest atomic number. In the example above, the substituent A is NH2 because N has a higher mass than C or H, the other atoms directly bounds to the assymetric carbon. To determine B, there are two substituents bound via one carbon and one via one hydrogen. As the mass of C is larger than the one of H, we look at the substituents of the carbons, in the methyl group, there are 3H and in the ethyl there is two H and one C, so the ethyl group primes over the methyl group. We have thus A=NH2, B=C2H5, C=CH3 and D=H. The next step is to place the liaison between the carbon and the substituent of lowest atomic number in our axis of sight. The carbon is between us and the substituent D. The three other substituents are pointing towards us and are approximatively in the plane of the papersheet. We can draw the liaisons between them and the carbon.
If A, D and C are in the horlogic order, the carbon is R. Otherwise it is S.
The second method uses the representation of Fisher on which the four liaisons are on the same plane. In reality it is not the case: to obtain this representation, imagine that you hold any two substituents with your thumb and your forefinger (for instance B and C).
Then you rotate the molecule to form an horizontal plane with the assymetric carbon, pointing in your direction. The other two liaisons are vertical and pointing away from you. The goal now is to place the substituent of lowest priority at the top of the representation. To do that, we can swap the places of the substituents if the are direct neighbours. It is almost as simple as that: for each permutation made, the configuration also changes from R to S or vice versa. As a result, if we made two permutations, the configuration remains the same (R→S→R). We apply this at the end, but remember the number of permutations made.
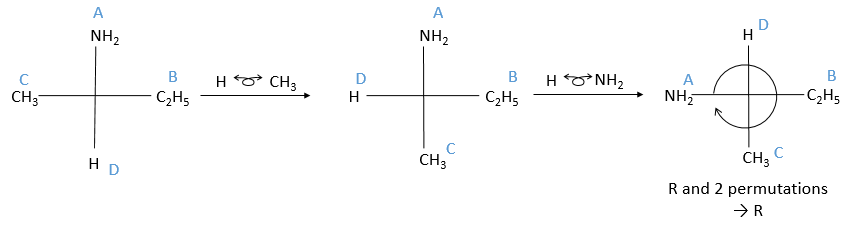
When the lowest priority subsitutent is at the top, we ignore it and we determine the “observed configuration” the same way than in the previous method: if they are in the horlogic order, then it is R, otherwise it is S. To obtain the real configuration from the observed configuration we just determined, we swap between R and S for each permutation made.
You may want to place the substituents in the horlogic order with permutation in all cases. You can do it if it helps you, but don’t forget that each permutation changes the configuration.
2) Axis of chirality
A similar method is followed. The difference is that we separate the substituents by their side of the axis ‘noted with and without prime). We will use the projection of Newton following the axis of chirality. The substituents at the front (the side we put in front does not matter) are noted a and b in function of their atomic number and the ones at the back a’ and b’. Next we check if aba’ is in the horlogic order (aR) or not (aS).
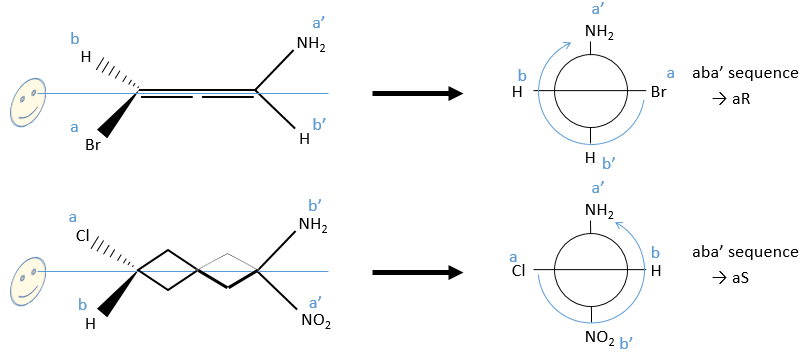
3) Helicoidal chirality
If the helix goes from the top to the bottom in the horlogic order, then it is noted P or (+). Otherwise it is M or (-).
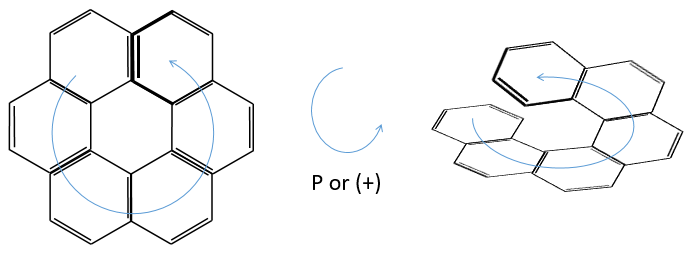
Diastereoisomery
Two stereoisomers that are not enantiomers are diastereoisomers. It can be the case if there are several stereogenic centres in the molecule.
Two stereogenic centres
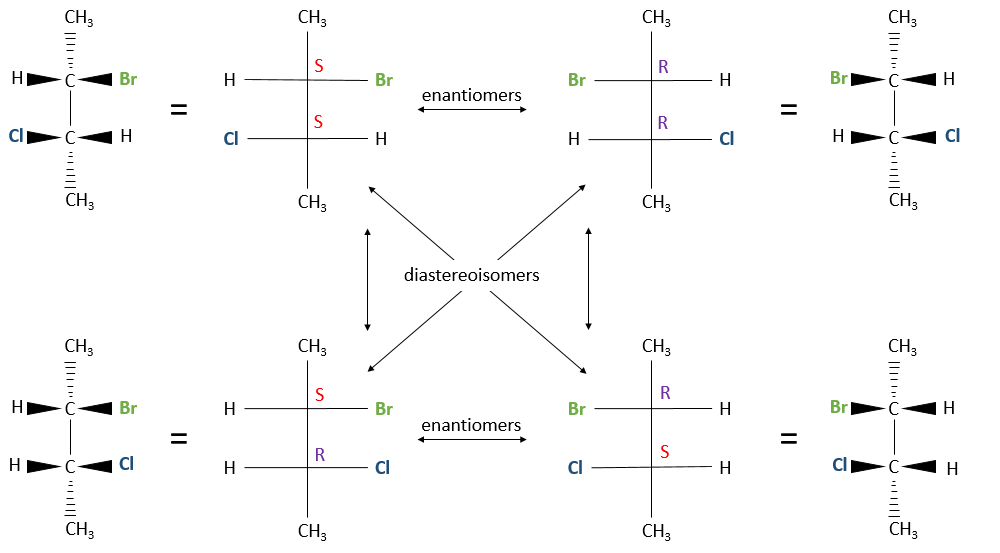
When the configurations are identical (RR or SS) are called like and when they are different (RS or SR), called unlike. Diastereoisomers haven’t the same chemical and physical properties because the interatomic distances are different. In the previous figure for instance, the distance between Br and Cl is different for the RS and the RR configurations. It is thus possible to separate them with usual techniques.
In the case of two asymmetric carbons, there is one enantiomer by configuration and two diastereoisomers. Yet the two diastereoisomers are sometimes one unique configuration that we call the meso form. For instance there is only one diastereoisomer for the enantiomers (2R,3R) and (2S,3S) of the 2,3-butanediol.
The reason here is a plane of symmetry between the carbon 2 and 3 of the butanediol.
Double liaisons
The presence of a double liaison entraves the rotation of the liaison. If there is at least one different substituent on each of the two carbons, there are then two stereoisomers that are called Z is the substituents of largest atomic number are in cis position, i.e. on the same side of the double liaison, and E if the substituents are in trans position, i.e. on the opposite side of the double liaison.
Prochirality
An achiral molecule is said to be prochiral if when we replace one of the substituent by a different one it becomes chiral. It implies that the two ligands that were apparently indiscernable are not equivalent.
Homotopy and heterotopy
In a not-prochiral molecule, the substituents that are identicals and are related one each other by a rotation around an axis of symmetry of order n are said to be homotopic or equivalent. If we replace one of those equivalent substituent by a different one, the product is identical than if we replaced the other homotopic substituent.
In opposition, we talk about eniantiotopic ligands (or substituents) if there was a centre of prochirality. The two ligands are identical and are the result of a symmetry of each other but if we replace one of them by another substituent, then we don’t get the same molecule than if we replaced the other eniantiotopic ligand.
To name these molecules, we use the CIP method and we randomly give the priority to one of the eniantiotopic ligand. This ligand is pro-R if the CIP method gives R and pro-S if the CIP method gives S. The second eniantiotopic ligand has the opposite name.
Two eniantiotopic ligands can’t be distinguished by usual physical and chemical methods, by RMN for instance. However, a chiral reactant like an enzyme can make the difference between the pro-R and pro-S ligands.
Eniantiotopic faces
In addition to eniantiotopic ligands, we also talk about eniantiotopic faces if a double liason is bound to a prochiral centre. The face of the plane on which the substituents are in the horlogic order is called face re and the other face is called face si.
The probabilities to attack by the Re or by the Si face are equal if the nucleophilic species is achiral. In this case we obtain a 50/50 racemic melange of the enantiomers R and S. If the nucleophile is chiral, then the reactions on the Re and Si faces have different speeds. The reaction is diastereoselective.
Diastereotopic ligands and faces
A prochiral centre can be near an asymmetric carbon. Two identical ligands of the prochiral centre are then not equivalent because they are not at the same distance from some groups of the asymmetric carbon. The replacement of one of the ligands or of the other leads to two diastereoisomers.
Achiral reactants can make the distinction between diastereotopic faces, one being usually more hindred than the other one. It leads to one product more formed than the other one. The products are diastereoisomers.
The less hindred side is usually favoured (model of Felkin).
Racemisation
It is the passage from one active optical body to the corresponding racemic melange. The racemisation can be due to time, heat or chemicals. In a general way, the cleavage of one liaison is involved and a carbon in the intermediate species possesses one sp2 carbon.
An example of racemisation is the enol-ketone tautomery. Starting from a solution made of one single enantiomer of a ketone, the other enantiomer will appear if there is a trace of a base in the solution because of the passage by the enol form. Under the sp2 form, the carbon is plane and there is thus no more S or R configuration. Both enantiomers can be formed from the enol.
Another example is the passage from one enantiomer to the other by the formation of a carbocation.
Epimerisation
Epimers are diastereoisomers with several stereogenic centres that differ only by the configuration of one of those centres. The epimerisation is the passage from one epimer to the other one.
Chapter 2 : chemical kinetics – catalysis
Homogeneous catalysis
In some conditions, a same reaction A → B can lead to two different kinetics depending on the composition of the solution in which it is. One species C, apparently not intervening in the reaction as it is not consumed by it, increases the speed of reaction.
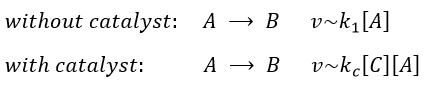
Both reactions occur simultaneously, giving a global reaction speed of
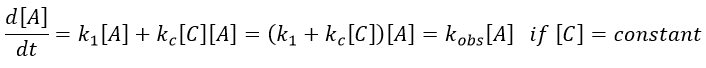
The ratio between the two paths is given by kc[C]/k1. The concentration of the catalyst can thus affect the contribution of the reaction paths. If the product of the reaction is the catalyst, then we say that the reaction is autocatalytic: the presence of the product enhances its own formation.
The use of a catalyst doesn’t change the difference of enthalpy and entropy between the products and the reactants but only the reaction path. The catalysed reactions involves several steps of smaller energy of activation than the normal reaction
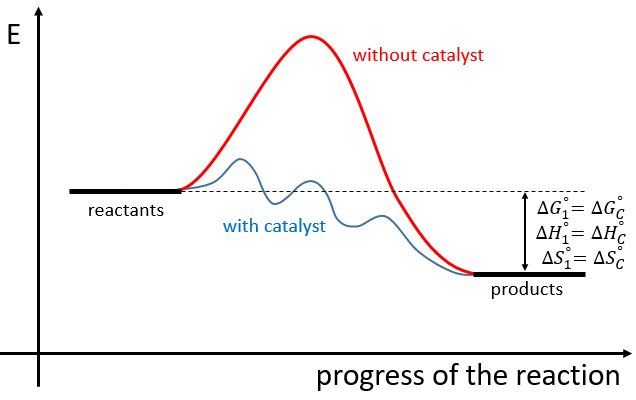
If the reaction is a reaction of equilibrium, the catalyst is catalyst of both sides of the reaction.
Electrophile catalysis – example of the hydrolysis of an ester
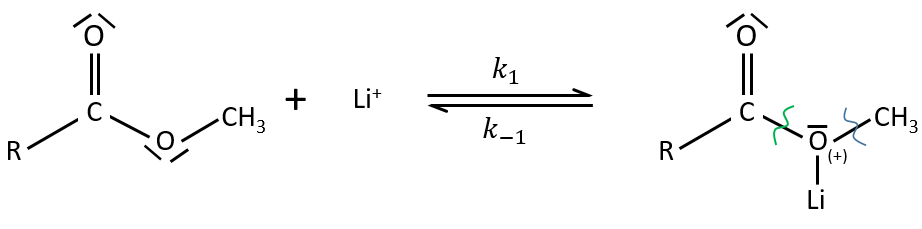
The steric hindrance can make the hydrolysis difficult. In this case we can use anhydric
LiI/pyridine to catalyse the reaction. Li+ is an electrophile that is complexed by the -O-. It acts as an acid of Lewis. The complexation eases the departure of R’ (-CH3 in the present case) which is normally a bad leaving group. K+ or Na+ can also be used.
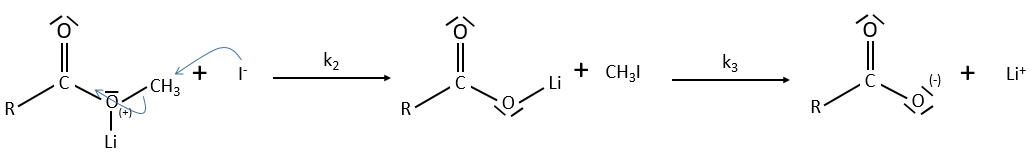
Specific acid catalysis
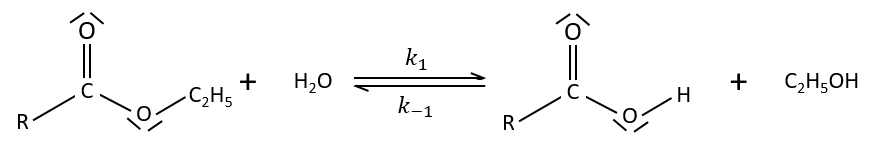
The formation of an acid A from an ester E is very slow in absence of catalyst. Proton can play the role of the catalyst in this reaction. The speed of the reaction will thus depend on the pH of the solution. The source of proton doesn’t matter but H3O+ is the catalyst.
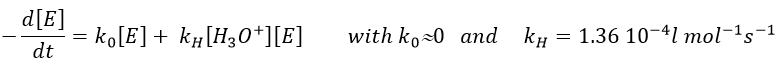
The reaction is reversible but we measure a pseudo order 1 if the acid is diluted: there is almost no product and the reverse reaction is thus negligible.
The concentration of the dissociated acid is calculated by the usual acid-base equation.
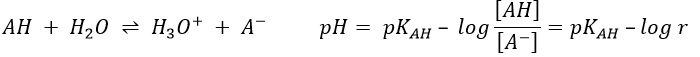
If [AH] varies at a constant pH, r is constant and thus kobs doesn’t depend on the concentration of AH. We are in the case of a specific acid catalysis.

The hydrolysis depends thus strongly on the acid-base equilibrium. The mechanism is
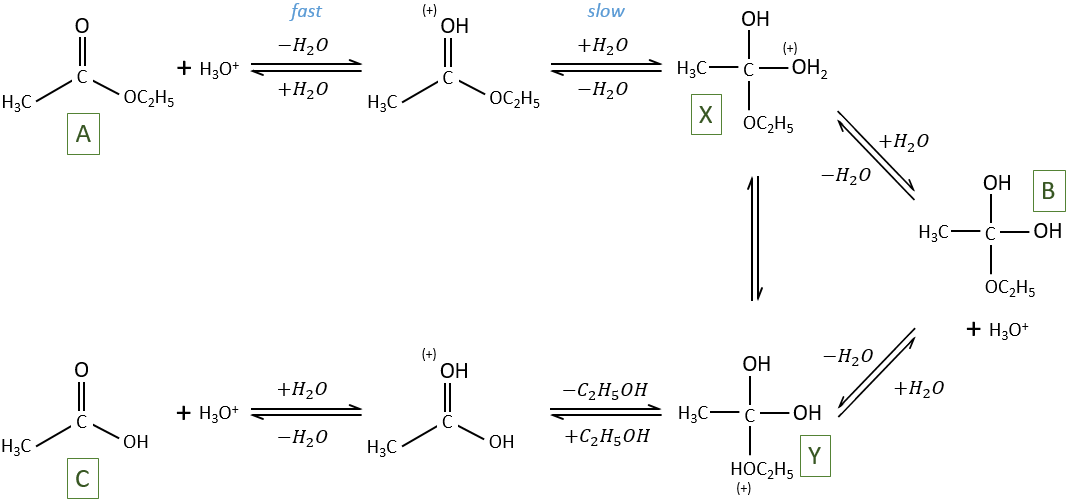
The protonation (first reaction) is fast and the nucleophilic attack by H2O (second reaction) is slow. X can turn into Y directly or first into B and then into Y.
General acid catalysis
An undissociated weak acid can also catalyse the reaction. If we have [H3O+] and [HA] constant, then
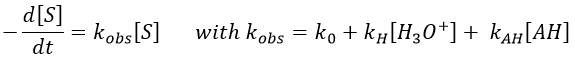
For a constant ratio [AH]/[A–], kobs varies with the concentration of [AH]. If there is a reaction involving AH, we have to recalculate [H3O+]. If there is no AH in solution, then we are back in the case of a specific acid catalysis.
Example of the vinyl ether
The formation of an aldehyde and an alcohol from a vinyl ether (noted V) is catalysed by an acid. In a neutral or basic environment, the vinyl ether is stable (k0»0).

The protons are much more effective than the weak acid and it can be seen from the intensity of the constants of speed. For instance, if R=C2H5, kH= 2.08l mol-1s-1 and kacetic acid= 1.78 10-3 l mol-1s-1.
Comparison between specific and general acid catalysis: aldehyde into acetal reaction
In this case there is a difference of mechanism between the general and specific acid catalyses.
Specific:
Protons are directly involved in the first step, so the pH has an influence on the speed of reaction.
General:
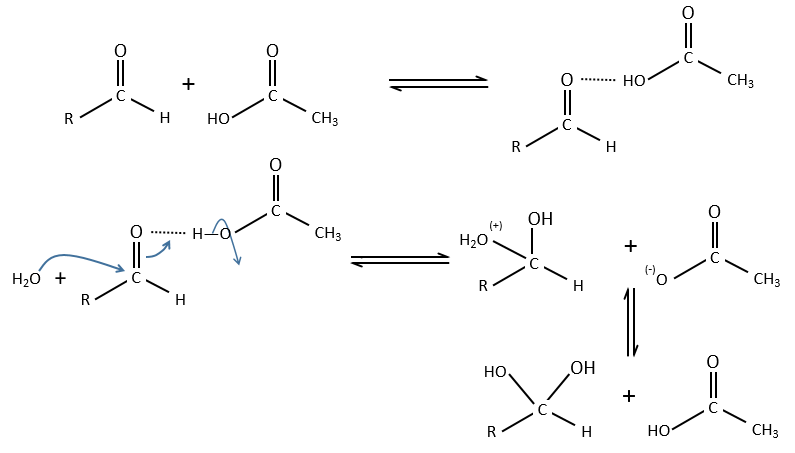
A weak acid forms an H bond to enhance the electrophile character of the carbon of the C=O and the transfer of proton is done simultaneously with the nucleophilic attack by the water. No proton are involved so the pH doesn’t change the dynamics. So to determine which catalysis is used, we change the concentration of the weak acid without touching the pH.
Nucleophilic catalysis
The catalyst interacts with an electrophile centre of the substrate. The process is slower and the intermediate species can be detected by spectroscopy or can be chemically trapped.
Example of the pyridine as catalyst
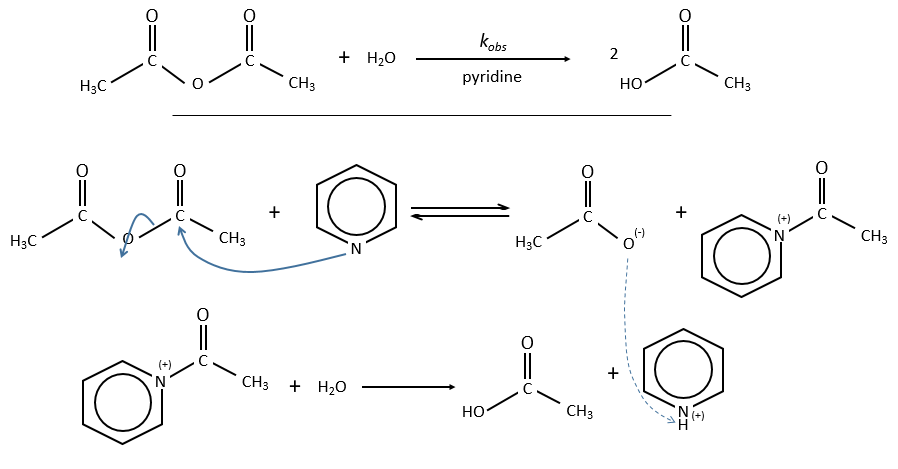
The acylpyridinium has a half-life time of 0.1s and can thus be detected.
Basic catalysis
Difference between specific and general basic catalyses
Specific:
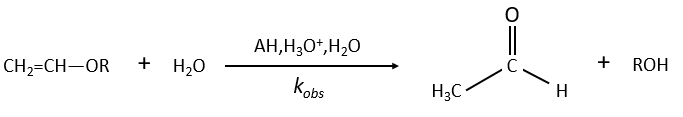
General:
The NR3-H2O complex is more reactive than water because the lone pair of the oxygen is more nucleophilic.
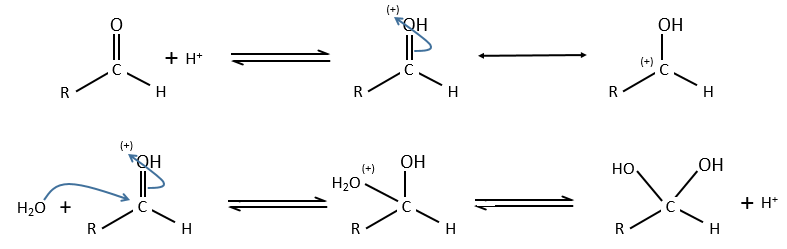
OH– does not directly intervene in the general basic catalysis, as H3O+ does in the general acid catalysis. Yet the constant of speed depends on the pH because the general and specific catalysis are simultaneous. The general (basic or acid) catalyses are thus an important mechanism at pH’s close to 7, for instance in enzymatic reactions.
Enzymatic catalysis
It can be done several ways
- catalysis by proximity (entropic contribution)
- electrophile and nucleophile catalyses
- general basic catalysis
Catalysis by proximity
We compare here an intramolecular process with an intermolecular process.
Intermolecular
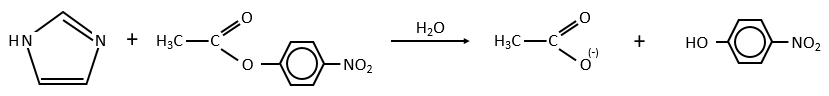
Imidazole is incorporated into many important biological molecules. One of them is the histidine that is present in many proteins and enzymes. The presented reaction is of the second order.
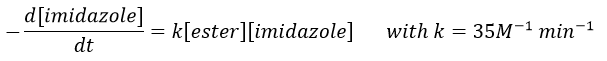
The p-nitrophenol is in equilibrium with its deprotonated form at neutral pH, form that can be detected via spectrometry of absorption.
Intramolecular
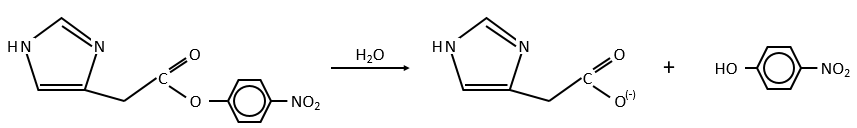
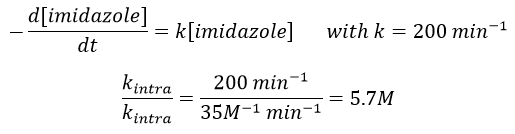
The fact that the catalyst was initially bound to the reactant is thus equivalent to the presence of 5.7M of the ester. The fact that the catalyst is already well positioned plays a role and there is also an entropic factor: from one molecule we produce two molecules and there is no need to bind the reactant to the catalyst anymore.
Nucleophilic catalysis
There are nucleophilic groups on enzymes. They can thus act as nucleophile catalysts (see previously).
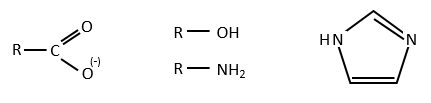
Equation of Michaelis-Menten
The enzymes are in small quantity in comparison to their substrates. The enzyme forms a complex with its substrate and then acts on it to form the product.
From the section on an equilibrium followed by an irreversible reaction, we know that
In this section, we have seen that there are two limit cases: the irreversible reaction is very fast or the equilibrium has time to be reached. In this case, the equilibrium has time to be established.
The quantity of enzyme is conserved over the time, in its lone form or complexed with the substrate.
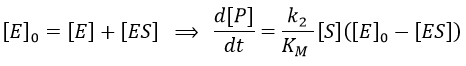
We used the hypothesis of stationarity on [ES].
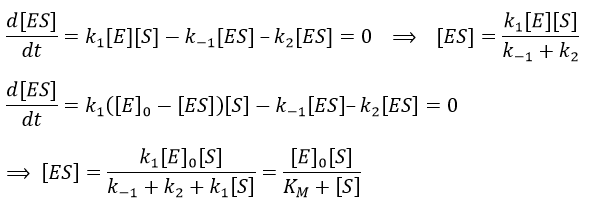
The formation of the product P is given by
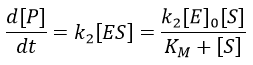
The speed of the reaction depends thus strongly on the concentration of the substrate up to a given point where [S]>>KM. The speed reaches then a plateau at v=k2[E]0. The maximum of the speed is thus reached when all the enzymes are active simultaneously ([ES]=[E]0).
![]()
The half of this speed is reached for a concentration [S]=KM.
By plotting 1/v vs 1/[S] (before the plateau), we find a straight line from which we can determine KM and k2.
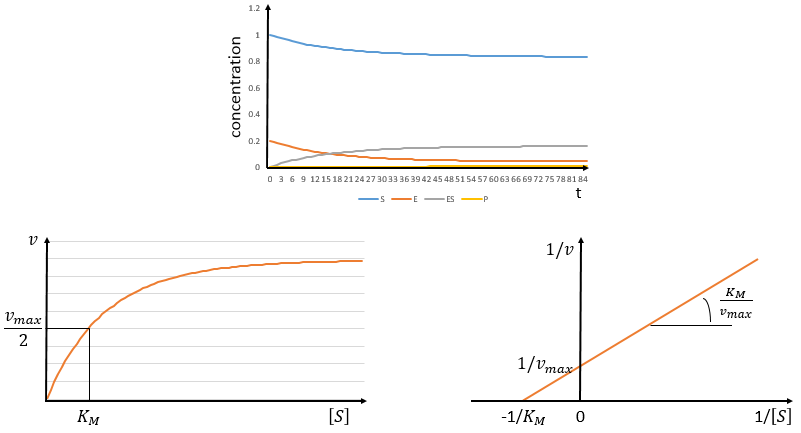
If we consider one more step in the reaction, the formation of the product when it is still complexed with the enzyme, i.e. before the separation of the species, we will have a correction factor γ to add.
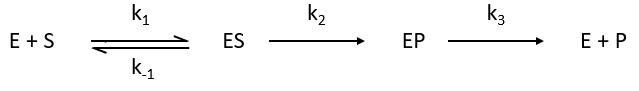
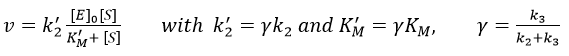
Case of the α-chymotrypsin
This enzyme is active in the digestive system where it hydrolyses polypeptides. It is active in this basic environment (pH between 8 and 9) and breaks the amide bonds between the peptides.
For the experiments, we substitute the polypeptides by an ester, the p-nitrophenylacetate.
The p-nitrophenol is detected due to its yellow colour (405nm) and the acetate is not detected. The observations of the experiment are particular: the enzyme acts very fast at the beginning of the experiment and then the speed reaches a constant level. It is thus the opposite of what we have seen just above. If we look at the small times, the production of phenol is directly proportional to the quantity of enzyme. It seems that it is in this step of the reaction that we have vmax=k2[E]0. There should be a step after the release of the phenol that inhibits the enzyme. This step is the release of the acetate which is much slower than the release of the phenol.
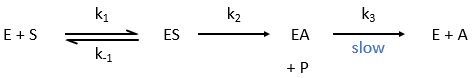
where P=p-nitrophenol, A=phenylacetate and k3<<k2.
As a result, when the phenol is released, the enzyme is not yet available to cleave another substrate. It has first to release the acetate, which is much slower. At the beginning of the reaction, we observe an important and quick increase of [phenol] that is quickly released by the enzyme. Before this moment, all the enzymes can do their job, what explains the large speed of reaction. After, one fraction of the enzymes is still occupied by one fragment of the substrate and cannot react until the acetate is released. The speed of the reaction is thus limited by the third step of the reaction.
We can assume that there is a nucleophilic site on the enzyme where the substrate binds at the level of the carbonyl.
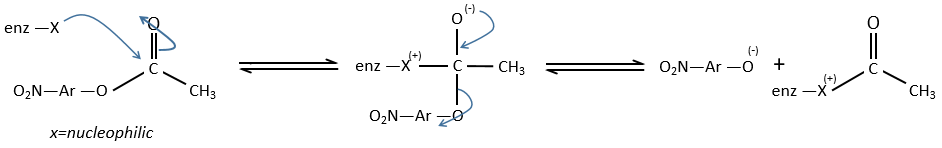
We can show that the intermediate acylenzyme is correct with the use of a more hindered substrate.
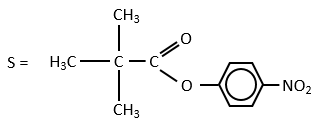
The steric hindrance and the inductive character of the methyl’s slow down the reaction, and thus they increase the life time of the acyled intermediate (~200 minutes). We can isolate the intermediate as crystals and analyse them.
We can use a better nucleophile than water to eliminate the acetate faster.