Dans ce chapitre nous allons discuter des types de réactions qui peuvent être effectuées sur les composés organiques. Dans ce cours de la première année nous nous concentrerons uniquement sur l’une des réactions les plus utilisés : la substitution nucléophile (SN).
La réaction de substitution est en effet la base de la chimie organique : son but est de remplacer un groupe d’une molécule donnée par une autre désirée. D’autres types de réactions sont l’addition et l’élimination (ils seront vus dans les cours de deuxième année).

A la fin nous pouvons, en théorie, faire ce que nous voulons avec les composés organiques. Pour l’instant vous avez déjà la moitié de l’explication du nom de la substitution nucléophile.
Un nucléophile est un composé, riche en électrons, qui est attiré par le noyau (pauvre en électrons) ainsi ils peuvent partager une paire de ces électrons pour former une liaison. Par opposition un électrophile est un composé, pauvre en électrons, qui est attiré par les électrons. D’autre part l’acidité, comme nous l’avons vu précédemment, n’est pas importante dans ces réactions. L’acidité est importante dans des solutions aqueuses mais les composés organiques ne se dissolvent pas bien dans une solution aqueuse. Dans les solutions aqueuses les espèces sont dissociées alors que dans les solutions organiques elles ne le sont pas. Les procédés de séparation tirent parti de cela pour purifier des composés distincts.
Lors d’une substitution nucléophile un nucléophile attaque une molécule cible pour substituer un groupe appelé groupe partant (LG). La cible de l’agent nucléophile est un atome de carbone pauvre en électrons car il appartient à un groupe d’atome plus électronégatif. En général les cibles sont limitées aux halogènes et aux groupes O ou N (les groupes nitro n’en font pas partie parce que l’azote a une charge positive dans ce cas). Dans le cas de SN le groupe partant prend une paire d’électrons avec lui.
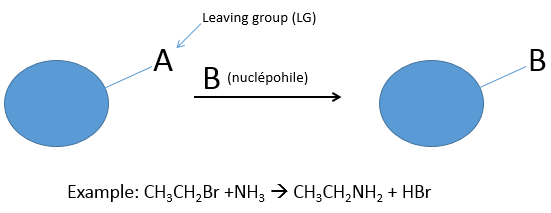
Le groupe partant est un nucléophile lui-même et pour que la réaction soit effectuée il faut que le nucléophile attaquant soit plus fort que le groupe partant. Plusieurs paramètres sont déterminants dans la force nucléophile (ou nucléophilie).
Charge du nucléophile
Tout d’abord un nucléophile n’a pas à être chargé, le fait de posséder un doublet libre d’électrons est suffisant. H2O, NH3, CH3OH sont tous de bons nucléophiles grâce à leurs paires d’électrons libres (sur le O et sur le N). Cela dit, leur équivalent chargé négativement est un nucléophile plus fort.
![]()
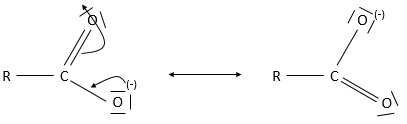
Soit la charge ou la paire d’électrons libres doivent être disponibles pour la réaction. Si la charge (ou la paire libre) est délocalisée la nucléophilie diminue de manière significative. Malgré leurs paires libres et leurs charges les carboxylates ne sont pas de bons nucléophiles parce que leurs charges peuvent être déplacées. On dit qu’un carboxylate est stabilisé par résonance parce que ce composé a une tendance moindre à réagir avec d’autres composés en raison de ses deux formes de résonance.
Basicité
Même si l’acidité est moins pertinente dans des solutions organiques, il existe une bonne relation entre la basicité d’un composé et sa force nucléophile.
L’électronégativité
La nucléophilie décroît avec l’électronégativité de l’atome portant la charge/ doublet. Rappelez-vous que l’électronégativité est la capacité de l’atome de garder ses électrons. Par conséquent l’atome électronégatif ne partagera pas ses électrons pour former une liaison.
Polarisabilité
nucléophilie décroît avec la polarisabilité de l’atome. Alors qu’il est contre-intuitif en ce qui concerne la réaction, ce rapport est dû à la solvatation de l’agent nucléophile. Dans les solvants protiques les espèces nucléophiles sont entourées par les molécules de solvant en formant des liaisons hydrogène avec les doublets libres de l’agent nucléophile. Fait intéressant la solvatation est plus forte pour les petites molécules que pour les plus grandes parce que la densité de charge est plus grande pour les petites molécules.
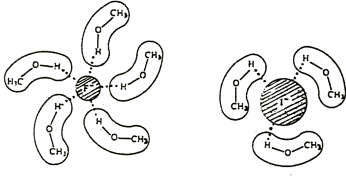
Dans les solvants aprotiques cet effet n’existe pas et la polarisabilité favorise la nucléophilie.
Mécanismes de substitutions nucléophiles :
Il existe 3 mécanismes possibles pour une substitution :
– tout d’abord l’attaque du substrat par l’agent nucléophile puis l’éjection du groupe partant
– attaque simultanée du nucléophile et le départ du groupe partant.
– tout d’abord le départ du groupe partant et ensuite l’attaque par le nucléophile.
Le premier mécanisme doit rapidement être ignoré car la première étape mène à un atome de carbone pentavalent. Les deux autres mécanismes existent et sont en concurrence. Dans certaines conditions les deux peuvent se produire en même temps. Nous allons maintenant voir les mécanismes en détail et les différences entre eux.
SN2 : Substitution nucléophile bimoléculaire
SN2 est le deuxième mécanisme de la liste : attaque simultanée du nucléophile et le départ du groupe partant. L’ensemble du processus est effectué en une seule étape et la vitesse de la réaction dépend de la concentration des deux réactifs (substrat et le nucléophile). C’est la raison pour laquelle ce mécanisme est appelé bimoléculaire.
Notre première étape est de déterminer où le nucléophile attaque sur le substrat. Imaginez la substitution d’un atome de chlore par un alcool sur une molécule donnée (X, Y et Z étant le reste de la molécule), le nucléophile (OH-) a deux options :
Attaquer par le côté du chlore ou par l’autre côté
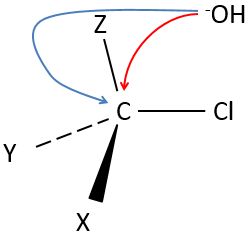
Les deux parties donnent le même résultat dans sa composition mais la connectivité diffère.
L’ attaque par le côté du groupe partant (appelé attaque frontale)
Pour être simultanée la liaison entre le substrat et le groupe partant desserre alors que la liaison entre le substrat et le nucléophile se forme. Il donne lieu à un complexe transitoire pour obtenir ensuite l’alcool substitué.

La connectivité de la molécule formée est identique à celle de la molécule de départ : X, Y et Z ne changent pas de position pendant le processus.
L’attaque par l’autre côté (appelé attaque dorsale)
Encore une fois un complexe transitoire est formé. Ce complexe montre des différences en ce qui concerne le complexe transitoire observé par une attaque frontale : la conformation de la molécule change au cours du processus. Les liaisons entre X, Y et Z avec C sont dans un plan, perpendiculaire au plan des «liaisons» avec OH et Cl ..
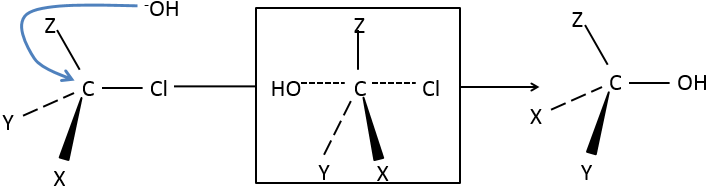
En conséquence, les positions de X et Y sont inversées. Les produits des substitutions frontale et dorsale sont des produits chiraux (images en miroir l’un de l’autre).
Nous pouvons donc facilement déterminer quels énantiomères sont obtenus en utilisant l’activité optique de chaque énantiomère. Elle a montré que seulement la substitution dorsale est produite. L’attaque frontale ne se produit donc pas.
Il ya deux conséquences :
– il ya une inversion de configuration après une SN2.
– l’encombrement stérique ralentit ou bloque la réaction. La taille de X, Y et Z joue donc un rôle important dans la vitesse de la réaction. Par exemple la SN2 est 145 fois plus rapide pour CH3Br que pour CH3CH2Br qui lui-même, est 128 fois plus rapide que pour (CH3).
SN1 : substitution nucléophile monomoléculaire
Dans ce mécanisme le départ du groupe partant intervient en premier et ensuite le nucléophile attaque le substrat.
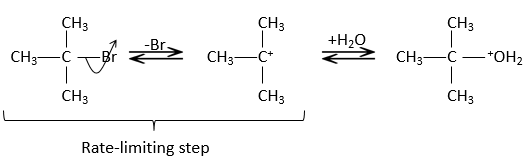
Il y a donc deux étapes dans ce mécanisme. La vitesse globale de la réaction est donnée par l’étape la plus lente appelé l’étape limitante ou étape cinétiquement déterminante qui est le départ du groupe partant. En effet, elle implique la rupture d’une liaison et une séparation des charges. Un solvant protique aide à séparer le groupe partant et le carbocation (cation carbonique) par solvatation des espèces chargées. La deuxième étape est beaucoup plus rapide : le nucléophile est attiré par ce carbone portant la charge positive.
L’étape limitante est donc la première étape de la substitution où le groupe partant est dissocié du substrat. Cette réaction implique un seul réactif le nucléophile agit uniquement à la deuxième étape de la substitution. Le nom de SN1, substitution nucléophile monomoléculaire vient de là. La cinétique dépend alors uniquement de la concentration du substrat contrairement à la SN2 où la concentration du nucléophile a une influence sur la vitesse de réaction. Une façon de déterminer si une réaction de substitution est SN1 ou SN2 est d’effectuer une analyse de la cinétique de la réaction et de voir si la concentration du nucléophile a ou n’a pas d’influence sur la vitesse de réaction.
Pour avoir une SN1 le carbone chargé positivement, appelé carbocation, doit être stabilisé.
Carbocation
Un carbocation est un carbone portant une charge positive. Il dispose de trois liaisons dans un seul plan et une orbitale vide perpendiculaire à ce plan.
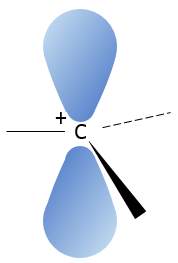
Cette configuration géométrique stabilise le carbocation avec des angles optimisés entre les liaisons (nb : une structure en forme d’avion ou quasi avion doit être obtenu) mais aussi par hyperconjugaison. Hyperconjugaison est l’interaction des électrons dans une liaison sigma (généralement C-H ou C-C) avec une orbitale p vide non liant adjacente (ou partiellement remplie) (le cas de carbocations), orbitale σ ou π antiliante ou orbitale π rempli pour donner une orbitale moléculaire étendue qui augmente la stabilité du système. En d’autres termes les liaisons des atomes de carbone adjacents aux carbocations stabilisent les carbocations lorsqu’elles sont orientées correctement.
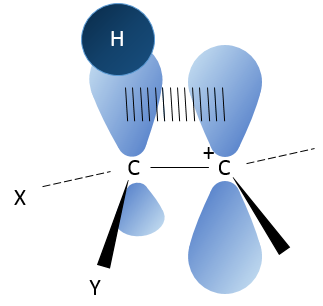
Dans l’image ci-dessus on peut voir l’hyperconjugaison d’une liaison sp entre C-H avec l’orbitale p vide du carbocation.
En conséquence les carbocations substitués sont stabilisées par hyperconjugaison. La séquence de stabilité pour carbocations est donc :
![]()
Comme vous avez peut-être déjà compris un paramètre pour déterminer si le mécanisme de substitution est un SN1 ou SN2 est la substitution du carbone portant le groupe partant. Des atomes de carbone trisubstitués conduisent à SN1 et les carbones monosubstitués ou nonsubstitués conduisent à SN2. Avec les carbones bisubstitués les deux mécanismes peuvent prendre place et sont assez lents.
Les liaisons π adjacentes les stabilisent également par (hyper) conjugaison des orbitales et par résonance.
![]()
Revenons au mécanisme de SN1. Les carbocations sont planes et peuvent être attaqués par les deux côtés du plan. Les proportions d’attaque de chaque côté sont égales sauf s’il existe une différence d’encombrement stérique pour un côté.
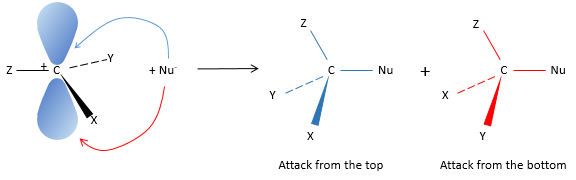
À la suite de la SN1, on obtient un mélange racémique des deux énantiomères.
Cependant nous pouvons parfois obtenir des produits très différents et inattendus d’un SN1. Par exemple :
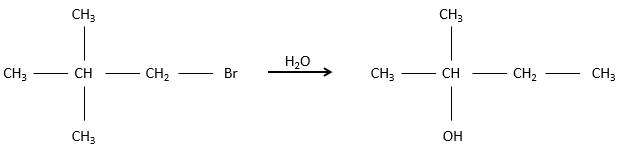
ce produit est tout à fait inattendu : le groupe partant n’a pas été substitué par un nucléophile mais plutôt par un groupe méthyle tandis que le nucléophile a pris la place d’un groupe méthyle même pas sur le même carbone que le groupe partant. La raison pour laquelle un tel produit est obtenu est que le carbocation produit au cours de la SN1, n’a pas été le carbocation le plus stable que la molécule pourrait avoir. Grâce à un mécanisme, pas totalement compris (appelé mécanisme de Wagner-Meerwein), un groupe de migrants (un méthyle ou un hydrogène) est déplacé pour obtenir une meilleure carbocation :
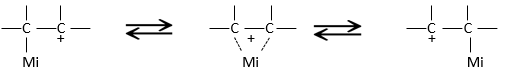
si un groupe méthyle et un hydrogène étaient sur le carbone adjacent, l’hydrogène est déplacée car elle conduit à une meilleure carbocation.

Seulement alors le nucléophile attaque le carbocation.
SN1 vs SN2
Les mécanismes eux-mêmes ne sont pas compliqués, mais il peut être difficile de déterminer quel mécanisme a lieu. Nous pouvons reprendre les paramètres influençant le mécanisme de substitution comme suit :
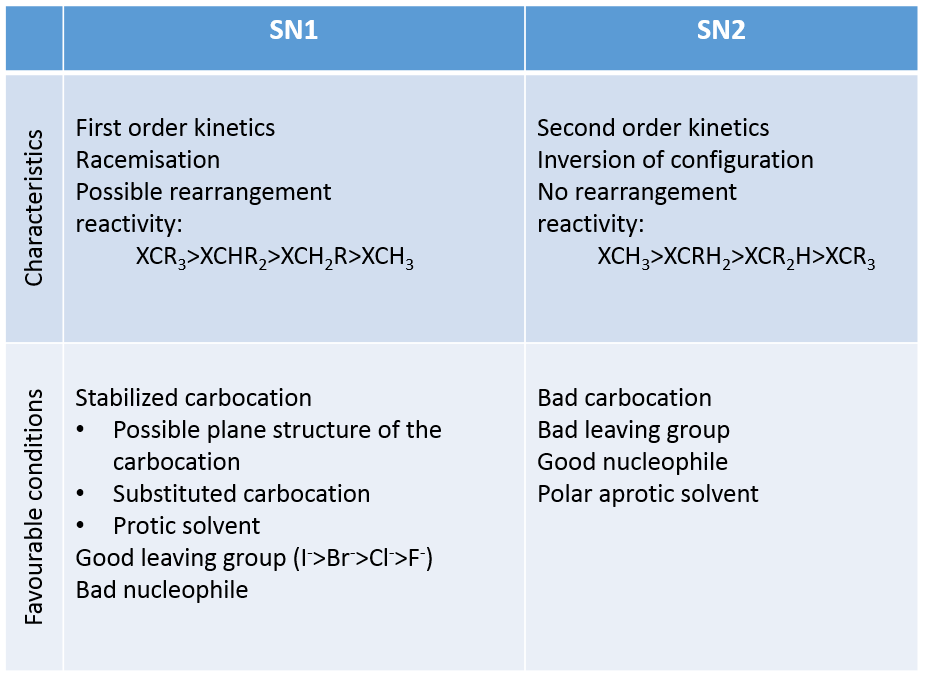
Comme il a été dit au début de ce chapitre, les substitutions nucléophiles sont l’une des réactions les plus importantes dans la chimie organique. Fondamentalement vous aurez envie de remplacer un groupe pour étendre une chaîne, de placer un groupe désiré sur le substrat ou de protéger un groupe.
D’autres substitutions que SN1 et SN2 existent mais nous les verrons ultérieurement.