Vecteurs: la Sélection et réplication autonome de l’ ADN :
Le clonage d’un morceau d’ADN exige qu’il soit répliqué quand il est remis dans les cellules. D’où l’ADN à cloner doit lui-même être une unité répliquante indépendante, un réplicon ou doit être joint à un réplicon. De plus, étant donné que l’efficacité de l’introduction de l’ADN dans des cellules est bien inférieur à 100%, les cellules qui ont absorbé l’ADN et on dit qu’elles ont été transformées, doivent être facilement identifiables. En effet, étant donné que seulement une bactérie cellule sur 105 est transformée, les sélections doivent généralement être incluses pour autoriser uniquement les cellules transformées de croître. Les vecteurs doivent remplir les deux conditions décrites ci-dessus, la réplication dans la cellule hôte et la sélection des cellules ayant reçu l’ADN transformant. Comme mentionné précédemment deux principaux types de vecteurs sont utilisés : des plasmides et des phages. Les plasmides contiennent des réplicons bactériens qui peuvent coexister avec l’ADN cellulaire normal et au moins un gène sélectionnable. Habituellement c’est un gène conférant une résistance à une antibiotique. Les phages contiennent bien sûr des gènes pour la réplication de leur ADN. Comme l’ADN emballé dans une enveloppe de phage peut pénétrer dans les cellules de manière efficace des gènes sélectionnables sur les phages habituellement ne sont pas nécessaires.
Vecteur plasmidique :
La plupart des plasmides sont de petits cercles qui contiennent les éléments nécessaires pour la réplication de l’ADN, un ou deux gènes de résistance aux médicaments et une région d’ADN dans laquelle l’ADN étranger peut être inséré sans endommager les fonctions essentielles du plasmide.
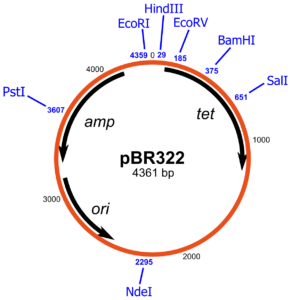
Un plasmide largement utilisé, pBR322, porte des gènes codant pour la résistance à la tetracycline et β-lactamase celle-ci confère une résistance à la pénicilline et aux analogues apparentés en clivant les médicaments dans le cycle lactame, ce qui les rend biologiquement inactifs. Des gènes conférant une résistance au chloramphénicol, à la tétracycline et kanamycine sont d’autres marqueurs de résistance aux médicaments couramment sélectionnables portés sur des plasmides.
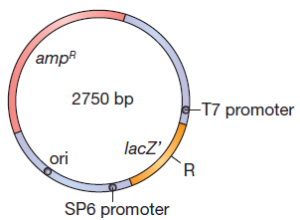
Un élément utile à avoir sur les plasmides est une origine de réplication de l’ADN à partir d’un phage simple brin. Quand une telle origine est activée par l’infection de tel phage, la cellule synthétise des quantités importantes d’un seul brin de l’ADN du plasmide. Ceci facilite le séquençage de l’ADN. Dans une expérience typique de clonage un plasmide est coupé dans une région non essentielle par une enzyme de restriction par exemple EcoRI et l’ADN monocaténaire étranger, coupé par EcoRI, est ajouté et les extrémités hybrides sont soudées ensemble. Seule une petite fraction des plasmides, soumis à ce traitement contiendra l’ADN inséré. La plupart auront recircularisé sans insertion d’ADN étranger. Comment les bactéries transformées c.à.d ceux dont les plasmides contiennent l’ADN inséré peuvent être distinguées des autres (ceux dont le plasmide reste sans ADN inséré)? Bien entendu dans certaines conditions une sélection génétique peut être utilisée pour activer uniquement la croissance des éléments transformés avec le fragment souhaité d’ADN inséré. Le plus souvent cela n’est pas possible et il devient nécessaire d’identifier des candidats qui contiennent l’ADN inséré.Une méthode pour identifier des candidats repose sur l’inactivation par l’ insertion d’un gène de résistance aux médicaments.
Par exemple, dans la résistance à l’ampicilline, le gène de la résistance sur pBR322 possède un seul site de clivage du plasmide par l’enzyme de restriction PstI. Heureusement le clivage par PstI génère des extrémités collantes et l’ADN peuvent être facilement soudées à ce niveau, après quoi il désactive le gène de la résistance à l’ampicilline. Le gène de résistance à la tétracycline sur le plasmide demeure intact et peut être utilisé pour la sélection des cellules transformées par le plasmide recombinant. Les colonies résultantes peuvent être testées en repérant sur une paire de plaques, l’une contenant de l’ampicilline, et l’autre sans ampicilline. Seul les mutants ampicilline sensibles, tétracycline-résistants vont contenir l’ADN étranger dans le plasmide alors que les ampicilline résistantess possèdent des plasmides re-circularisés sans insertion de l’ADN étranger.
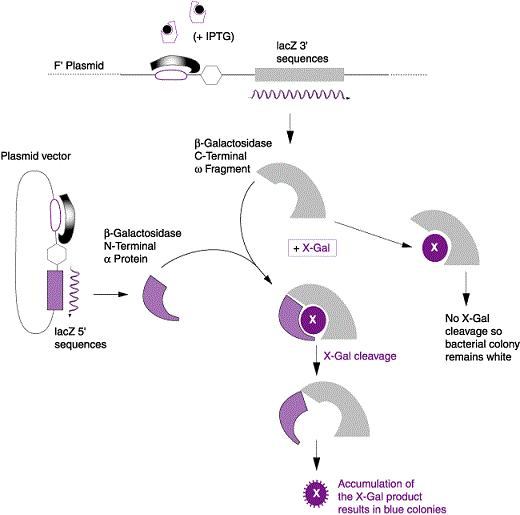
Une autre façon pour vérifier l’insertion d’ADN étranger, utilise le gène de ß-galactosidase. L’insertion d’ADN étranger dans le gène inactive l’enzyme qui peut être détectée en mettant les cellules transformées sur un milieu qui sélectionne la présence du plasmide et contient également des substrats de β-galactosidase qui produisent des colorants lorsqu’il hydrolyse ces substrats.
Un plasmide serait lourd s’il contenait la totalité des 3000 paires de bases du gène β galactosidase. Par conséquent seule une extrémité N-terminale du gène est placé sur le plasmide. Le reste de l’enzyme est codée par un segment inséré dans le chromosome des cellules hôtes. Les deux parties du gène synthétisent des domaines qui se lient entre eux pour donner l’enzyme actif. Ce phénomène inhabituel est appelé alpha-complémentation.
Des vecteurs de clonage sont conçus pour l’insertion d’ ADN étranger dans la partie N-terminale de la β-galactosidase. Une technique simple peut réduire considérablement la re-circularisation des molécules du vecteur sans insertion de l’ADN étranger. Si l’ADN du vecteur est traité par une phosphatase après coupure par l’enzyme de restriction, la re-circularisation devient impossible parce que l’extrémité 5′-PO4 requise pour l’ ADN ligase est absente. L’ADN étranger, cependant, contient une extrémité 5′-PO4 et par conséquent deux des quatre fragments d’ADN encadrant un fragment d’ADN étranger peuvent être soudés. Cet ADN est actif dans la transformation parce que les cellules réparent le pseudo restant à chaque extrémité du fragment inséré. Les vecteurs de clonage plasmides ou phages peuvent contenir un court tronçon de l’ADN contenant des sites de coupure unique pour plusieurs enzymes de restriction. Ces régions à liaisons multiples (polylinker) permettent le clivage par deux enzymes de sorte que les extrémités collantes résultantes ne sont pas auto-complémentaire. Donc le vecteur ne peut pas se re-circularisé et être resoudé sur lui-même mais quand un fragment d’ADN contenant les extrémités nécessaires et complémentaires aux extrémités du plasmide existe alors il peut le faire. Une génie génétique efficace nécessite que l’ADN plasmidique soit obtenu en grandes quantités. Certains plasmides maintiennent seulement trois ou quatre copies par cellule, tandis que d’autres plasmides ont 25 à 50 copies cellulaires. La plupart des plasmides ayant un nombre importants de copies peuvent être amplifiée du fait que le plasmide continue de se répliquer après que la synthèse protéique et la synthèse de l’ADN cellulaire ont cessé en raison d’une densité cellulaire élevée ou la présence d’inhibiteurs de la synthèse des protéines. Après amplification, une cellule contenant un tel plasmide peut contenir jusqu’à 3000 copies de plasmide.

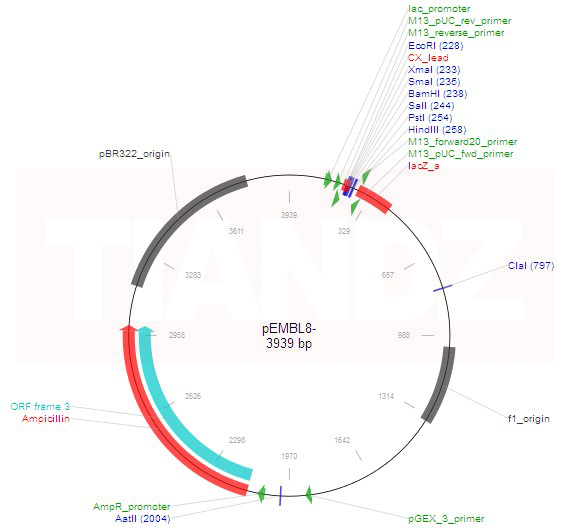
La nature n’a pas livré des vecteurs plasmidiques prêt pour le génie génétique.Les vecteurs plasmidiques les plus utiles ont été eux-mêmes construits par génie génétique. Les matières de départ sont les plasmides R, ce sont des plasmides ou des éléments d’ADN à réplication autonome qui portent un ou plusieurs gènes de résistance aux médicaments. Les plasmides R sont la cause de graves problèmes médicaux, car diverses bactéries peuvent acquérir des plasmides R et ainsi devenir résistant aux médicaments normalement utilisés pour le traitement des infections . La conversion d’un plasmide R en un vecteur utile nécessite l’élimination de l’ADN étranger et l’élimination des multiples sites de clivage par des enzymes de restriction. Afin que l’ADN étranger puisse être cloné dans le plasmide, le plasmide doit posséder un seul site de clivage pour au moins une enzyme de restriction, et cela devrait être dans une région non essentielle. Dans la construction des vecteurs de clonage, des plasmides ont été digérés avec diverses enzymes de restriction. Le mélange résultant de fragments d’ADN a été hybridés ensemble par l’intermédiaire des extrémités auto-complémentaires, puis collées pour produire de nombreuses combinaisons de fragments possibles . Ces ADN ont été transférés dans des cellules. Seuls les nouveaux plasmides contenant au moins les segments d’ADN nécessaires à la réplication et à la résistance aux médicaments ont survécu et ont donné des colonies. Les plasmides souhaitables contenant uniquement les sites de clivage uniques pour des enzymes de restriction peuvent être identifiés par l’amplification et la purification de l’ADN suivie par des digestions test avec des enzymes de restriction suivi d’électrophorèse pour caractériser les Produits de la digestion. Le plasmide pBR322 possède une seule site de clivage pour plus de 20 enzymes de restriction. Parmi les plus communément utilisés sont Bam HI, Eco RI, Hind III, PstI, PvuII et Sali.
Un vecteur phagique pour les bactéries :
des vecteurs de phage et des vecteurs dérivés de phages sont utiles pour trois raisons:
-les phages peuvent transporter de plus grands fragments d’ADN insérés que des plasmides. Donc beaucoup moins de candidats transformés doivent être examinés pour trouver un clone désiré.
-L’efficacité d’infecter les cellules par l’ADN des phages reconditionnés est considérablement plus grande que l’efficacité de la transformation avec L’ADN de plasmide dans les cellules. Ceci est un facteur important quand un clone rare est recherché.
-Enfin, le phage lambda permet un procédé commode pour le criblage pour détecter le clone porteur du gène désiré. Cependant une fois qu’un segment désiré d’ADN a été cloné dans un phage la commodité de manipuler des plasmides, en partie en raison de leur petite taille, dicte que le segment soit sous-cloné dans un plasmide.
Vecteur de clonage basé sur le bactériophage M13 :
L’exigence la plus essentielle pour tout vecteur de clonage est qu’il ait un moyen de se répliquer dans la cellule hôte. Pour les vecteurs plasmidiques, cette exigence est facile à satisfaire, car des séquences d’ADN relativement courtes sont capables d’agir comme origines de réplication plasmidiques, et la plupart, sinon la totalité, des enzymes nécessaires à la réplication sont fournies par la cellule hôte. Des manipulations élaborées, telles que celles qui ont abouti à pBR322, sont donc possibles tant que la construction finale a une origine de réplication fonctionnelle et intacte. Avec des bactériophages tels que M13 et e, la situation en matière de réplication est plus complexe. Les molécules d’ADN phagique portent généralement plusieurs gènes essentiels à la réplication, y compris des gènes codant pour des composants de la couche protéique du phage et des enzymes réplicatives spécifiques de l’ADN phagique. La modification ou la suppression de l’un de ces gènes altèrent ou détruisent la capacité de réplication de la molécule résultante. Il y a donc beaucoup moins de liberté pour modifier les molécules d’ADN phagique, et généralement les vecteurs de clonage de phages ne sont que légèrement différents de la molécule parente.
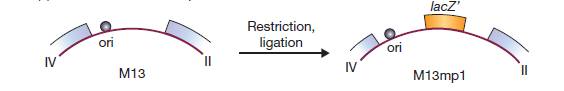
Les problèmes de construction d’un vecteur de phages sont illustrés en considérant M13. Le génome M13 normal a une longueur de 6,4 kb, mais la plus grande partie de ce génome est occupée par dix gènes étroitement liés, chacun étant essentiel à la réplication du phage. Il n’y a qu’une seule séquence intergénique de 507 nucléotides dans laquelle un nouvel ADN pourrait être inséré sans perturber l’un de ces gènes, et cette région comprend l’origine de réplication qui doit elle-même rester intacte. Clairement, il n’y a qu’une possibilité limitée de modifier le génome M13.
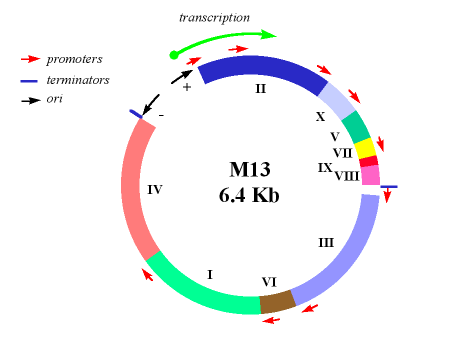
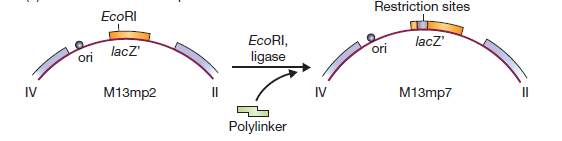
La première étape de construction d’un vecteur M13 consistait à introduire le gène lacZ ‘dans la séquence intergénique. Cela a donné M13mp1, qui forme des plaques bleues sur de l’agar X-gal.
M13mp1 ne possède pas de sites de restriction uniques dans le gène lacZ ‘. Cependant, il contient l’hexanucléotide GGATTC près du début du gène. Un seul changement de nucléotide rendrait ce GAATTC, qui est un site EcoRI.
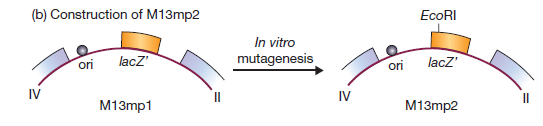
Cette altération a été réalisée en utilisant la mutagenèse in vitro, résultant en M13mp2
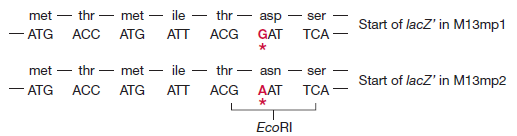
M13mp2 a un gène lacZ′ légèrement modifié (le sixième codon spécifie maintenant l’asparagine à la place de l’acide aspartique), mais l’enzyme β-galactosidase produite par les cellules infectées par M13mp2 est encore parfaitement fonctionnelle. L’étape suivante dans le développement des vecteurs M13 consistait à introduire des sites de restriction supplémentaires dans le gène lacZ’. Ceci a été réalisé en synthétisant dans le tube à essai un oligonucléotide court, appelé polylinker, qui consiste en une série de sites de restriction et a des extrémités collantes EcoRI.
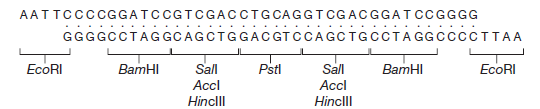
Ce linker multisite a été inséré dans le site EcoRI de M13mp2, pour donner M13mp7, un vecteur plus complexe avec quatre sites de clonage possibles (EcoRI, BamHI, Sali et PstI). Le polylinker est conçu pour ne pas perturber totalement le gène lacZ’: un cadre de lecture est maintenu dans tout le polylinker, et une enzyme β-galactosidase fonctionnelle bien que modifiée est encore produite. Les vecteurs M13 les plus sophistiqués ont des polylinkers plus complexes insérés dans le gène lacZ’. Un exemple est M13mp8, qui a la même série de sites de restriction que le plasmide pUC8 . Comme avec le vecteur plasmidique, un avantage de M13mp8 est sa capacité à prendre des fragments d’ADN avec deux extrémités cohésives différentes.
Phagemides (vecteurs hybrides formés de plasmides et M13) :
Bien que les vecteurs M13 soient très utiles pour la production de versions monocaténaires de gènes clonés, ils souffrent d’un inconvénient. Il y a une limite à la taille du fragment d’ADN qui peut être cloné avec un vecteur M13, avec 1500 pb comme la capacité maximale, bien que des fragments jusqu’à 3 kb ont parfois été cloné. Pour contourner ce problème, un certain nombre de vecteurs hybrides (« phagemides ») ont été développé en combinant une partie du génome M13 avec l’ADN plasmidique. Un exemple est fourni par pEMBL8, qui a été faite en transférant dans pUC8 un fragment de 1300 pb du génome M13.
Ce morceau d’ADN M13 contient la séquence signal reconnue par les enzymes qui convertissent le double brin normal de M13 en ADN simple brin avant la sécrétion de nouvelles particules phagiques. Cette séquence de signaux est toujours fonctionnelle même si elle est détachée du reste du génome de M13, les molécules pEMBL8 sont également converties en ADN simple brin et sécrété comme particules phagiques défectueuses.
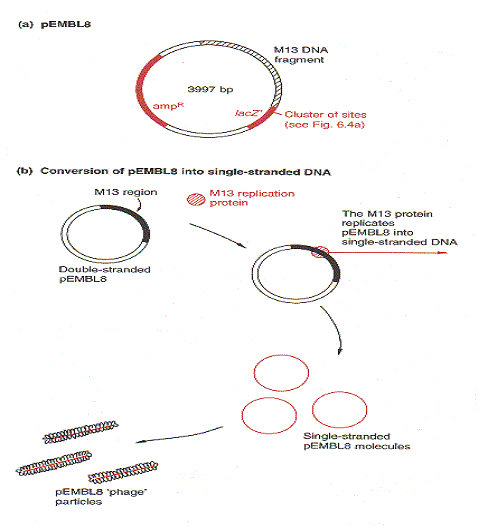
Tout ce qui est nécessaire, c’est que les cellules de E. coli utilisées en tant qu’hôtes pour une expérience de clonage pEMBL8 sont ensuite infectés avec M13 normal à agir comme un phage auxiliaire, fournissant les enzymes réplicatives nécessaires et les protéines de l’enveloppe du phage. pEMBL8, dérivé de pUC8, a les sites de clonage de polylinker dans le lacZ ‘ gène, de sorte que les plaques recombinantes peuvent être identifiées de manière standard sur de l’agar contenant X-gal. Avec pEMBL8, versions monocaténaires de fragments d’ADN clonés jusqu’à 10 kb en longueur peut être obtenue, en étendant considérablement la portée du système de clonage M13.
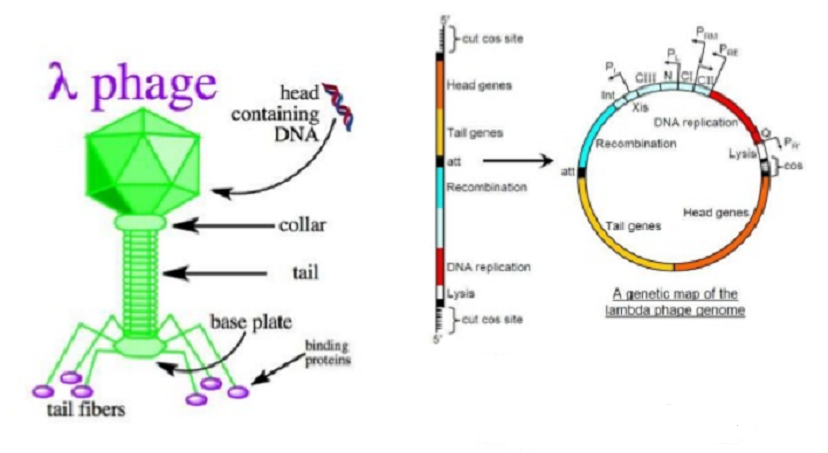
Vecteurs de clonage basés sur bactériophage λ :
Deux problèmes devaient être résolus avant que les vecteurs de clonage basés sur λ puissent être développés:
l) La molécule d’ADN de λ peut être augmentée seulement d’environ 5%, ce qui représente l’addition de seulement 3 kb de nouveaux ADN. Si la taille totale de la molécule est supérieure à 52 kb, alors elle ne pourra pas être empaquetée dans la structure de la tête e et les particules de phages infectieuses ne sont pas formées. Ceci limite sévèrement la taille d’un fragment d’ADN qui peut être inséré dans un vecteur λ non modifié.
2) Le génome λ est si grand qu’il a plus d’une séquence de reconnaissance pour pratiquement chaque endonucléase de restriction. La restriction ne peut pas être utilisée pour cliver la molécule λ normale de manière à permettre l’insertion d’un nouvel ADN, car la molécule serait coupée en plusieurs petits fragments qui seraient très peu susceptibles de reformer un génome λ viable lors de la religation. Au vu de ces difficultés, il est peut-être surprenant qu’une grande variété de vecteurs de clonage λ aient été développés, leur utilisation principale étant de cloner de gros morceaux d’ADN de 5 à 25 kb, beaucoup trop gros pour être manipulés par des vecteurs plasmidiques ou M13.
Des segments du génome λ peuvent être supprimés sans compromettre sa viabilité :
La voie à suivre pour le développement de vecteurs de clonage a été fournie par la découverte qu’un grand segment dans la région centrale de la molécule d’ADN peut être éliminé sans affecter la capacité du phage à infecter des cellules de E. coli. L’élimination de tout ou partie de cette région non essentielle, jusqu’à 15 kb. Cela signifie que jusqu’à 18 kb de nouvel ADN peuvent maintenant être ajoutés avant que le seuil de l’emballage ne soit atteint . Cette région « non-essentielle » contient en effet la plupart des gènes impliqués dans l’intégration et l’excision du prophage λ du chromosome de E. coli. Un génome de λ modifié par suppression est donc non lysogène et ne peut suivre que le cycle d’infection lytique. Ceci est en soi souhaitable pour un vecteur de clonage car cela signifie qu’une induction n’est pas nécessaire avant la formation de plaques .
La sélection naturelle peut être utilisée pour isoler des λ modifiés dépourvus de certains sites de restriction :
Natural selection can be used to isolate modified * that lack certain restriction sites Even a deleted e genome, with the non-essential region removed, has multiple recognition sites for most restriction endonucleases. This is a problem that is often encountered when a new vector is being developed. If just one or two sites need to be removed, then the technique of in vitro mutagenesis (p. 200) can be used. For example, an EcoRI site, GAATTC, could be changed to GGATTC, which is not recognized by the enzyme. However, in vitro mutagenesis was in its infancy when the first e vectors were under development, and even today would not be an efficient means of changing more than a few sites in a single molecule. Instead, natural selection was used to provide strains of e that lack the unwanted restriction sites. Natural selection can be brought into play by using as a host an E. coli strain that produces EcoRI. Most e DNA molecules that invade the cell are destroyed
by this restriction endonuclease, but a few survive and produce plaques. These are mutant phages, from which one or more EcoRI sites have been lost spontaneously (Figure 6.11). Several cycles of infection will eventually result in e molecules that lack all or most of the EcoRI sites.
6.3.3 Insertion and replacement vectors
Once the problems posed by packaging constraints and by the multiple restriction sites had been solved, the way was open for the development of different types of λ-based cloning vectors. The first two classes of vector to be produced were λ insertion and λ replacement (or substitution) vectors.
λ Insertion vectors
With an insertion vector (Figure 6.12a), a large segment of the non-essential region has
been deleted, and the two arms ligated together. An insertion vector possesses at least
one unique restriction site into which new DNA can be inserted. The size of the DNA
fragment that an individual vector can carry depends, of course, on the extent to which
the non-essential region has been deleted. Two popular insertion vectors are:
l Egt10 (Figure 6.12b), which can carry up to 8 kb of new DNA, inserted into a
unique EcoRI site located in the cI gene. Insertional inactivation of this gene means
that recombinants are distinguished as clear rather than turbid plaques (p. 83).
λ replacement vectors.
A e replacement vector has two recognition sites for the restriction endonuclease used
for cloning. These sites flank a segment of DNA that is replaced by the DNA to be
cloned (Figure 6.13a). Often the replaceable fragment (or “stuffer fragment” in cloning
jargon) carries additional restriction sites that can be used to cut it up into small pieces,
so that its own re-insertion during a cloning experiment is very unlikely. Replacement
vectors are generally designed to carry larger pieces of DNA than insertion vectors can
handle. Recombinant selection is often on the basis of size, with non-recombinant vectors
being too small to be packaged into e phage heads (p. 84).
An example of a replacement vectors is:
l EEMBL4 (Figure 6.13b) can carry up to 20 kb of inserted DNA by replacing a
segment flanked by pairs of EcoRI, BamHI, and SalI sites. Any of these three
restriction endonucleases can be used to remove the stuffer fragment, so DNA
fragments with a variety of sticky ends can be cloned. Recombinant selection with
eEMBL4 can be on the basis of size, or can utilize the Spi phenotype (p. 83).
6.3.4 Cloning experiments with * insertion or replacement
vectors
A cloning experiment with a e vector can proceed along the same lines as with a plasmid
vector—the e molecules are restricted, new DNA is added, the mixture is ligated,
and the resulting molecules used to transfect a competent E. coli host (Figure 6.14a).
This type of experiment requires that the vector be in its circular form, with the cos
sites hydrogen bonded to each other.
Although satisfactory for many purposes, a procedure based on transfection is not
particularly efficient. A greater number of recombinants will be obtained if one or two
refinements are introduced. The first is to use the linear form of the vector. When the
linear form of the vector is digested with the relevant restriction endonuclease, the
left and right arms are released as separate fragments. A recombinant molecule can
100 Part I The Basic Principles of Gene Cloning and DNA Analysis
λ arms
EcoRI EcoRI
EcoRI
EcoRI
New DNA
cos cos
cos
cos cos
Catenane cos
EcoRI EcoRI
EcoRI
EcoRI
EcoRI, ligate
(a) Cloning with circular λ DNA
(b) Cloning with linear λ DNA
λ insertion factor
– circular form
cos cos
Transfect
E. coli
Infect E. coli
in vitro
packaging mix
Recombinant
molecule
Recombinant λ
New DNA
Ligate
Figure 6.14
Different strategies for cloning with a λ vector. (a) Using the circular form of λ as a plasmid. (b) Using left and right arms
of the λ genome, plus in vitro packaging, to achieve a greater number of recombinant plaques.
be constructed by mixing together the DNA to be cloned with the vector arms
(Figure 6.14b). Ligation results in several molecular arrangements, including catenanes
comprising left arm–DNA–right arm repeated many times (Figure 6.14b). If the inserted
DNA is the correct size, then the cos sites that separate these structures will be the right
distance apart for in vitro packaging (p. 81). Recombinant phage are therefore
produced in the test tube and can be used to infect an E. coli culture. This strategy, in particular
the use of in vitro packaging, results in a large number of recombinant plaques.
6.3.5 Long DNA fragments can be cloned using a cosmid
The final and most sophisticated type of e-based vector is the cosmid. Cosmids are
hybrids between a phage DNA molecule and a bacterial plasmid, and their design
centers on the fact that the enzymes that package the e DNA molecule into the phage
protein coat need only the cos sites in order to function (p. 21). The in vitro packaging
reaction works not only with e genomes, but also with any molecule that carries cos sites
separated by 37–52 kb of DNA.
A cosmid is basically a plasmid that carries a cos site (Figure 6.15a). It also needs a
selectable marker, such as the ampicillin resistance gene, and a plasmid origin of replication,
as cosmids lack all the e genes and so do not produce plaques. Instead colonies are
formed on selective media, just as with a plasmid vector.
Chapter 6 Cloning Vectors for E. coli 101
BamHI
ori
cos
BamHI
cos
Circular
pJB8
Restrict with
BamHI
BamHI BamHI
cos
cos cos
Linear pJB8 BamHI
BamHI BamHI BamHI
BamHI
New DNA
New
DNA
Ligate
In vitro package
Recombinant
cosmid DNA
Colonies containing circular
recombinant pJB8 molecules
Ampicillin medium
Infect E. coli
(a) A typical cosmid
(b) Cloning with pJB8
pJB8
5.4 kb
λ DNA
Catenane
λ particles
ampR
ampR
ampR
ampR
Figure 6.15
A typical cosmid and the way it is used to clone long fragments of DNA.
A cloning experiment with a cosmid is carried out as follows (Figure 6.15b). The
cosmid is opened at its unique restriction site and new DNA fragments inserted. These
fragments are usually produced by partial digestion with a restriction endonuclease, as
total digestion almost invariably results in fragments that are too small to be cloned with
a cosmid. Ligation is carried out so that catenanes are formed. Providing the inserted
DNA is the right size, in vitro packaging cleaves the cos sites and places the recombinant
cosmids in mature phage particles. These e phage are then used to infect an E. coli
culture, though of course plaques are not formed. Instead, infected cells are plated onto
a selective medium and antibiotic-resistant colonies are grown. All colonies are recombinants,
as non-recombinant linear cosmids are too small to be packaged into e heads.
6.4 8 and other high-capacity vectors enable
genomic libraries to be constructed
The main use of all e-based vectors is to clone DNA fragments that are too long to be
handled by plasmid or M13 vectors. A replacement vector, such as eEMBL4, can carry
up to 20 kb of new DNA, and some cosmids can manage fragments up to 40 kb. This
102 Part I The Basic Principles of Gene Cloning and DNA Analysis
Table 6.1
Number of clones needed for genomic libraries of a variety of organisms.
NUMBER OF CLONES*
SPECIES GENOME SIZE (bp) 17 kb FRAGMENTS† 35 kb FRAGMENTS‡
E. coli 4.6 × 106 820 410
Saccharomyces cerevisiae 1.8 × 107 3225 1500
Drosophila melanogaster 1.2 × 108 21,500 10,000
Rice 5.7 × 108 100,000 49,000
Human 3.2 × 109 564,000 274,000
Frog 2.3 × 1010 4,053,000 1,969,000
*Calculated for a probability ( p) of 95% that any particular gene will be present in the library.
†Fragments suitable for a replacement vector such as yEMBL4.
‡Fragments suitable for a cosmid.
compares with a maximum insert size of about 8 kb for most plasmids and less than 3 kb
for M13 vectors.
The ability to clone such long DNA fragments means that genomic libraries can be
generated. A genomic library is a set of recombinant clones that contains all of the DNA
present in an individual organism. An E. coli genomic library, for example, contains all
the E. coli genes, so any desired gene can be withdrawn from the library and studied.
Genomic libraries can be retained for many years, and propagated so that copies can be
sent from one research group to another.
The big question is how many clones are needed for a genomic library? The answer
can be calculated with the formula:
N =
where N is the number of clones that are required, p is probability that any given gene
will be present, a is the average size of the DNA fragments inserted into the vector, and
b is the total size of the genome.
Table 6.1 shows the number of clones needed for genomic libraries of a variety of
organisms, constructed using a e replacement vector or a cosmid. For humans and other
mammals, several hundred thousand clones are required. It is by no means impossible
to obtain several hundred thousand clones, and the methods used to identify a clone
carrying a desired gene (Chapter 8) can be adapted to handle such large numbers, so
genomic libraries of these sizes are by no means unreasonable. However, ways of reducing
the number of clones needed for mammalian genomic libraries are continually being
sought.
One solution is to develop new cloning vectors able to handle longer DNA inserts.
The most popular of these vectors are bacterial artificial chromosomes (BACs), which
are based on the F plasmid (p. 16). The F plasmid is relatively large and vectors derived
from it have a higher capacity than normal plasmid vectors. BACs can handle DNA
inserts up to 300 kb in size, reducing the size of the human genomic library to just
30,000 clones. Other high-capacity vectors have been constructed from bacteriophage
P1, which has the advantage over e of being able to squeeze 110 kb of DNA into its
ln(1 − p)
ln
A
1 − aD
C bF
Chapter 6 Cloning Vectors for E. coli 103
FURTHER READING
Further reading
Bolivar, F., Rodriguez, R.L., Green, P.J. et al. (1977) Construction and characterization of
new cloning vectors. II. A multipurpose cloning system. Gene, 2, 95–113. [pBR322.]
Frischauf, A.-M., Lehrach, H., Poustka, A. & Murray, N. (1983) Lambda replacement
vectors carrying polylinker sequences. Journal of Molecular Biology, 170, 827–842.
[The lEMBL vectors.]
Iouannou, P.A., Amemiya, C.T., Garnes, J. et al. (1994) P1-derived vector for the propagation
of large human DNA fragments. Nature Genetics, 6, 84–89.
Melton, D.A., Krieg, P.A., Rebagliati, M.R., Maniatis, T., Zinn, K. & Green, M.R. (1984)
Efficient in vitro synthesis of biologically active RNA and RNA hybridization probes from
plasmids containing a bacteriophage SP6 promoter. Nucleic Acids Research, 12,
7035–7056. [RNA synthesis from DNA cloned in a plasmid such as pGEM3Z.]
Sanger, F., Coulson, A.R., Barrell, B.G. et al. (1980) Cloning in single-stranded bacteriophage
as an aid to rapid DNA sequencing. Journal of Molecular Biology, 143, 161–178.
[M13 vectors.]
Shiyuza, H., Birren, B., Kim, U.J. et al. (1992) Cloning and stable maintenance of 300
kilobase-pair fragments of human DNA in Escherichia coli using an F-factor-based
vector. Proceedings of the National Academy of Sciences of the USA, 89, 8794–8797.
[The first description of a BAC.]
Sternberg, N. (1992) Cloning high molecular weight DNA fragments by the bacteriophage
P1 system. Trends in Genetics, 8, 11–16.
Yanisch-Perron, C., Vieira, J. & Messing, J. (1985) Improved M13 phage cloning vectors
and host strains: nucleotide sequences of the M13mp18 and pUC19 vectors. Gene,
33, 103–119.
capsid structure. Cosmid-type vectors based on P1 have been designed and used to clone
DNA fragments ranging in size from 75 to 100 kb. Vectors that combine the features
of P1 vectors and BACs, called P1-derived artificial chromosomes (PACs), also have a
capacity of up to 300 kb.
6.5 Vectors for other bacteria
Cloning vectors have also been developed for several other species of bacteria, including
Streptomyces, Bacillus, and Pseudomonas. Some of these vectors are based on
plasmids specific to the host organism, and some on broad host range plasmids able to
replicate in a variety of bacterial hosts. A few are derived from bacteriophages specific
to these organisms. Antibiotic resistance genes are generally used as the selectable
markers. Most of these vectors are very similar to E. coli vectors in terms of their general
purposes and uses.
Le phage Lambda était un choix idéal pour un vecteur de phage, car il était bien compris et facile à travailler. Plus important encore, le phage contient une région non essentielle interne vaste flanquée par des sites de clivage Eco RI. Par conséquent, cette région non essentielle peut être enlevée et l’ADN étranger inséré. Avant de l’ADN lambda coupé par Eco RI peut être utilisé pour le clonage, il était nécessaire d’éliminer les sites de clivage supplémentaires qui sont situés à dans les régions essentielles du génome lambda. Tout d’abord, un phage lambda hybride a été construit par recombinaison génétique in vivo. Cela manquait trois sites de clivage EcoRI à 0,438, 0,538 et 0,654 dans la région non essentielle mais a conservé les deux sites dans les régions essentielles. Ensuite, les deux autres les sites de clivage Eco RI ont été éliminés par mutation et sélection. Dans la sélection du phage ont été cyclée entre hôtes manquant et contenant les systèmes de modification de restriction EcoRI. Tous les phages avec le clivage des sites mutés de manière à être nonrecognizable par le système Eco RI aurait une plus grande probabilité d’échapper aux enzymes de restriction sur la croissance dans le second hôte. Après 10 à 20 cycles de cette sélection régime, Davis a trouvé un phage qui avait perdu l’un des deux sites R1, et après un 9 à 10 cycles supplémentaires, il a trouvé un mutant qui avait perdu la autre site. Ce phage mutant a ensuite été transformé en un vecteur de clonage utile par recombinaison avec le type sauvage lambda pour restaurer les trois EcoRI des sites de clivage situé dans la partie centrale du génome. L’ADN isolé des particules de phage lambda est linéaire et il peut être clivé par EcoRI pour donner les fragments centraux plus petits et les plus grands des bras gauche et droit. Les fragments d’ADN clonés pour être clivé par EcoRI peut ensuite être hybridé ensemble et ligaturé à droite purifié et bras gauche. Ce Soit l’ADN peut être utilisé tel quel pour transfecter des cellules rendues compétentes pour son prise ou il peut être encapsidé in vitro dans des têtes de phage et utilisé pour infecter
A J 2
Essentiel
b cI rouge int nin QSR
Essentiel
0,438 0,538 0,654 0,808 0,927
278 génie génétique et l’ADN recombinant
cellules. L’efficacité, par molécule de l’ADN, de l’emballage et l’infection est
beaucoup plus élevé que la transfection avec de l’ADN nu. Par conséquent, l’emballage est
utilisé lorsque le fragment à cloner est présent en quelques exemplaires seulement.
Vecteurs pour cellules supérieures
Le clonage de l’ADN dans des cellules plus élevées pose les mêmes problèmes que le clonage dans
bactéries. Les vecteurs doivent permettre simplement la purification de quantités considérables
de l’ADN, doit permettre la sélection des cellules transformées, et doit posséder
l’espace pour l’ADN inséré. Les vecteurs navettes, qui ont été largement
utilisé pour le clonage dans la levure, sont une solution élégante à ces exigences. Dans
plus de contenir les éléments de clonage de plasmide bactérien normal,
ils contiennent un réplicon de levure et un marqueur génétique sélectionnable dans la levure
(Fig. 9.10). En conséquence, de grandes quantités des vecteurs peuvent être obtenus
par croissance dans E. coli et ensuite transformée en levure. La capacité à
navette entre les bactéries et les levures permet d’économiser beaucoup de temps et de dépenses en
expériences de génie génétique.
Deux types d’origines de réplication de levure peuvent être utilisés dans la navette de levure
vecteurs. La première est une origine de réplication de l’ADN chromosomique de levure, également connue
comme un élément ARS. L’autre est l’origine 2 μ à partir des cercles. Ces
sont des éléments plasmidiques-like avec fonction inconnue qui se trouvent dans
Levure. Ils sont un peu plus stables que les vecteurs ARS. Nutritional
des marqueurs tels que l’uracile, l’histidine, la leucine et la biosynthèse du tryptophane
ont été utilisés comme gènes de sélection appropriée dans le auxotrophe
Levure.
Les virus constituent une base pour de nombreux vecteurs utiles dans plante supérieure et
des cellules animales. Par exemple, l’un des vecteurs les plus simples pour les mammifères
des cellules est le SV4O du virus simien. Il permet un grand nombre des mêmes clonage
que les opérations du phage lambda.
La terminologie utilisée avec des cellules de mammifères peut être déroutant.
« Transformation » peut signifier que les cellules ont reçu un plasmide. Ça peut
signifient également que les cellules ont perdu leur inhibition de contact. Dans cet état,
ils continuent de croître au-delà du stade de la monocouche de cellules confluentes à laquelle
ADN de levure
l’origine de replication
ampicilline
la résistance
tétracycline
la résistance
Origine de
Réplication de l’ADN
YRP17
TRP
E. coli
URA
ADN de levure
ADN de levure
Figure 9.10 La structure de
un vecteur pour la navette entre
E. coli et la levure. Il contient
les gènes qui permettent la réplication de l’ADN
et la sélection à la fois
organismes.
Vecteurs pour cellules supérieures 279
des cellules normales de mammifères cessent croissance. Transformation de la désinhibée
état de croissance peut résulter d’une infection par un virus de la tumeur comme causant
SV4O ou elle peut être le résultat d’une mutation du génome. Bien que la perte de
l’inhibition de contact pourrait être utile pour identifier les cellules qui ont incorporé
les hybrides SV4O ADN ou SV4O, cette propriété est d’une utilité limitée.
D’autres marqueurs génétiques sélectionnables convenables pour des cellules de mammifères sont
Obligatoire.
Un gène utile pour les sélections dans les cellules de mammifères a été le
le gène de la thymidine kinase, car les cellules TK + peuvent être sélectionnées en cultivant
elles dans un milieu contenant de l’hypoxanthine, de l’aminoptérine et de la thymidine.
A l’inverse, les cellules TK- peuvent être sélectionnées en les cultivant dans un milieu
contenant de la bromodésoxyuridine (Fig. 9.11). En outre, les virologues
ont découvert précédemment que les codes de virus de l’herpès simplex de son
propre thymidine kinase. Par conséquent, le génome viral peut être utilisé en tant que
source concentrée du gène sous une forme exprimable pour le clonage initial
expériences.
Bien que le gène de la thymidine kinase a été utile pour la sélection de cellules
qui ont absorbé l’ADN étranger, un gène de sélection qui ne nécessite pas
l’isolement préalable d’une thymidine kinase mutant négatif dans chaque cellule
ligne serait également utile. E. coli enzyme xanthine-guanine
Hypoxanthine + Aminopterin + thymidine
Précurseurs IMP XMP GMP
xanthine
(Voie faible
les cellules de mammifères)
xanthine phosphoribosyl
transférase
GTP
AMP
thymidine
kinase
Aminopterin
dUMP dTMP (BrdU-MP, toxiques)
mycophénolique
acide
Thymidine (BrdU)
hypoxanthine
Brdu TK
TK +
–
Sélectionner
Figure 9.11 Les voies métaboliques impliquées avec quelques gènes sélectionnables dans
les cellules de mammifères. IMP-inosine monophosphate, monophosphate XMP-xanthine,
GMP-guanosine monophosphate, duMP-désoxyuridine monophosphate,
dTMP-désoxythymidine monophosphate, Brdu-bromodésoxyuridine, Brdu-MP,
monophosphate bromodésoxyuridine. blocs aminoptérine tétrahydrofolate réductase,
qui est nécessaire pour la synthèse de l’IMP et dTMP et mycophénolique
l’acide bloque la synthèse de XMP.
280 génie génétique et l’ADN recombinant
gène transférase phosphoribosyl semble répondre à ces exigences.
Le produit protéique des fonctions de gènes dans des cellules de mammifères et
permet la croissance sélective des cellules non mutant qui contiennent l’enzyme
(Fig. 9.11). Le milieu de croissance requis contient la xanthine, l’hypoxanthine,
aminoptérine, et de l’acide mycophénolique. D’autres gènes dominants
utile pour la sélection des cellules transformées sont des mutants de dihydrofolate
réductase qui est résistante au methotrexate, un inhibiteur puissant de la
Enzyme de type sauvage, et la kanamycine-néomycine phosphotransférase. le
celle-ci est une enzyme dérivée d’un transposon bactérien et confère
résistance aux bactéries, des levures, des plantes et des cellules de mammifères à un composé
appelé G418. Bien entendu, pour une expression appropriée dans les cellules supérieures, la
gène doit être relié à une unité de transcription appropriée et doivent
contenir les signaux d’initiation et de polyadénylation traduction requis.
mettre l’ADN