La PCR est une technique très simple: tout ce qui se passe est qu’une petite région d’une molécule d’ADN, par exemple un seul gène, est copiée à plusieurs reprises par une enzyme ADN polymérase. Cela peut sembler un exercice plutôt trivial, mais il a une multitude d’applications dans la recherche génétique et dans des domaines plus larges de la biologie. Nous commencerons ce chapitre par un aperçu de la réaction en chaîne de la polymérase pour comprendre exactement ce qu’il réalise. Ensuite, nous examinerons les questions clés qui déterminent si une expérience de PCR individuelle est réussie, avant d’examiner quelques-unes des méthodes qui ont été conçues pour étudier les fragments d’ADN amplifiés qui sont obtenus.
La PCR (polymerase chaine reaction ) en détaille :
La réaction en chaîne par polymerase conduit à l’amplification sélective d’une région choisie d’une molécule d’ADN. Toute région de n’importe quelle molécule d’ADN peut être choisie, pour autant que les séquences aux frontières de la région soient connues. Les séquences de la frontière doivent être connues car, pour effectuer une PCR, deux oligonucleotides courts doivent s’hybrider à la molécule d’ADN, un à chaque brin de la double hélice . Ces oligonucleotides, qui servent d’amorces pour les réactions de synthèse d’ADN, délimitent la région qui sera amplifié. L’amplification est habituellement effectuée par l’enzyme ADN polymérase I de Thermus Aquatique. Cet organisme vit dans les sources chaudes, et bon nombre de ses enzymes, y compris la Taq polymerase, sont thermostables, ce qui signifie qu’ils sont résistants à la dénaturation par traitement thermique. Comme on le verra dans un instant, la thermostabilité de la polymérase Taq est une exigence essentielle dans la méthodologie PCR.
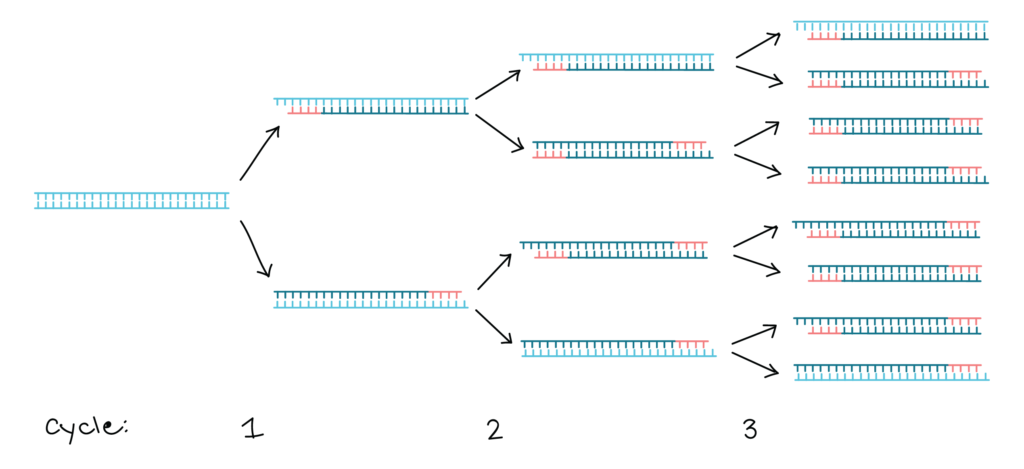
Pour réaliser une expérience de PCR, l’ADN cible est mélangé à la polymerase Taq, aux deux amorces oligonucléotidiques et à une réserve de nucleotides. La quantité d’ADN cible peut être très faible, car la PCR est extrêmement sensible et est capable de fonctionner avec une seule molécule de départ. On commence la réaction en chauffant le mélange à 94 ° C. A cette température, les liaisons hydrogène qui maintiennent ensemble les deux polynucleotides de la double hélice sont rompues, de sorte que l’ADN cible devient dénaturé en molécules monocaténaires. La température est ensuite réduite à 50-60 ° C, ce qui se traduit par une certaine réintégration des brins simples de l’ADN cible, mais permet également aux amorces de se fixer à leurs positions de recuit. La synthèse d’ADN peut maintenant commencer, de sorte que la température est élevée à 74 ° C, juste en dessous de l’optimum pour la polymerase Taq. Dans cette première étape de la PCR, un ensemble de « produits longs » est synthétisé à partir de chaque brin de l’ADN cible. Ces polynucleotides ont des extrémités 5 ‘identiques mais des extrémités 3’ aléatoires, ces dernières représentant des positions où la synthèse d’ADN se termine par hasard. Le cycle de dénaturation-recuit-synthèse est maintenant répété. Les produits longs dénaturés et les quatre brins résultants sont copiés au cours de la phase de synthèse d’ADN. Cela donne quatre molécules à double brin, dont deux sont identiques aux produits du premier cycle et dont deux sont entièrement faits d’ADN nouveau. Au cours du troisième cycle, ces derniers donnent lieu à des «produits courts» dont les extrémités 5 ‘et 3’ sont toutes deux fixées par les positions de recuit d’amorce. Dans les cycles suivants, le nombre de produits courts s’accumule de manière exponentielle (doublant à chaque cycle) jusqu’à ce que l’une des composantes de la réaction s’épuise. Cela signifie qu’après 30 cycles, il y aura plus de 130 millions de produits courts dérivés de chaque molécule de départ. En termes réels, cela équivaut à plusieurs microgrammes de produit de PCR à partir de quelques nanogrammes ou moins d’ADN cible.
A la fin d’une PCR, un échantillon du mélange réactionnel est habituellement analysé par l’électrophorèse sur gel de l’agarose , on verra qu’on a produit suffisamment d’ADN pour le fragment amplifié pour il soit visible sous la forme d’une bande discrète après coloration au bromure d’éthidium. Cela peut en soi fournir des informations utiles sur la région de l’ADN qui a été amplifiée ou, le produit de PCR peut être examiné par des techniques telles que le séquençage d’ADN.
Concevoir les amorces oligonucléotidiques pour une PCR:
Les amorces sont la clé du succès ou de l’échec d’une expérience PCR. Si les amorces sont conçues correctement, l’expérience conduit à l’amplification d’un seul fragment d’ADN, correspondant à la région cible de la molécule modèle. Si les amorces sont mal conçues, l’expérience échouera, peut-être parce qu’aucune amplification ne se produit, ou peut-être parce que le mauvais fragment ou plus d’un fragment est amplifié. De toute évidence, beaucoup de réflexion doit être placée dans la conception des amorces. L’élaboration de séquences appropriées pour les amorces n’est pas un problème: elles doivent correspondre aux séquences flanquant la région cible sur la molécule modèle.

Chaque amorce doit naturellement être complémentaire (non identique) à son brin matrice pour que l’hybridation se produise, et les extrémités 3 ‘des amorces hybridées doivent pointer l’une vers l’autre. Le fragment d’ADN à amplifier ne doit pas être supérieur à environ 3 kb de longueur et idéalement moins de 1 kb. Des fragments allant jusqu’à 10 kb peuvent être amplifiés par des techniques de PCR standard, mais plus le fragment est long, moins l’amplification est efficace et plus il est difficile d’obtenir des résultats cohérents. L’amplification de fragments très longs – jusqu’à 40 kb est possible, mais nécessite des méthodes spéciales. La première question importante à aborder est la longueur des amorces. Si les amorces sont trop courtes, elles pourraient s’hybrider à des sites non cibles et donner des produits d’amplification indésirables. Pour illustrer ce point, imaginez que l’ADN humain total est utilisé dans une expérience de PCR avec une paire d’amorces de huit nucléotides de longueur (dans le jargon de la PCR, on parle de «8-mères»). Le résultat probable est qu’un nombre de fragments différents seront amplifiés. En effet, on s’attend à ce que les sites de fixation de ces amorces se produisent, en moyenne, une fois tous les 48 = 65 536 pb, ce qui donne approximativement 49 000 sites possibles dans la séquence nucléotidique de 3 200 000 kb qui constitue le génome humain. Cela signifie qu’il serait très peu probable qu’une paire d’amorces 8-mères donnerait un seul produit d’amplification spécifique avec de l’ADN humain.
Que se passe-t-il si on utilise des amorces 17-mères ? La fréquence attendue d’une séquence 17-mères est une fois tous les 417 = 17 179 869 184 pb. Ce chiffre est plus de cinq fois supérieur à la longueur du génome humain, si bien qu’un amorce 17-mère devrait avoir un seul site d’hybridation dans l’ADN humain total. Une paire d’amorces 17-mer devrait donc donner un seul produit d’amplification spécifique . Pourquoi ne pas simplement faire les amorces aussi longtemps que possible? La longueur de l’amorce influe sur la vitesse à laquelle elle s’hybride à l’ADN matrice, les amorces longues s’hybridant à une vitesse plus lente. L’efficacité de la PCR, mesurée par le nombre de molécules amplifiées produites au cours de l’expérience, est donc réduite si les amorces sont trop longues, car une hybridation complète aux molécules matrices ne peut se produire dans le temps imparti pendant le cycle réactionnel. En pratique, les amorces plus longues que 30-mers sont rarement utilisées.
Working out the correct temperatures to use :
Au cours de chaque cycle de PCR, le mélange réactionnel est transféré entre trois températures:
1) La température de dénaturation, habituellement 94 ° C, qui casse les paires de bases et libère l’ADN monocaténaire pour servir de modèle pour la synthèse de nouvel ADN.
2) La température d’hybridation ou de recuit, à laquelle les amorces se fixent l’ADN:
La température de recuit (hybridation) est la plus importante car, encore une fois, cela peut affecter la spécificité de la réaction. L’hybridation ADN-ADN est un phénomène. Si la température est trop élevée, aucune hybridation n’a lieu et au lieu de cela les amorces et les brins modèles restent dissociés. Toutefois, si la température est trop basse et peu correspondante, dans lesquels toutes les paires de bases formés sont stables et aussi les mismatches. Si cela se produit, les calculs antérieurs concernant les longueurs appropriées pour les amorces deviennent non pertinentes, car ces calculs supposaient que seuls les hybrides parfaits d’amorce-modèle peuvent se former. Si des mismatches (inadéquations) sont tolérées, Le nombre de sites d’hybridation potentiels pour chaque amorce est grandement augmenté, et l’amplification est plus susceptible de se produire à des sites non cibles dans la molécule modèle.La température de recuit idéale doit être suffisamment faible pour permettre l’hybridation entre l’amorce et le model, mais suffisamment élevée pour empêcher les mismatches de former l’ADN. Cette température peut être estimée en déterminant la melting température (fusion) et ctte température ou la Tm de l’hybride amorce-matrice. La Tm est la température à laquelle l’hybride correctement formé se dissocie. Une température de 1-2 °C en dessous de cette température être suffisamment faible pour permettre la formation de l’hybride primaire-modèle correctement, mais elle est trop elevée pour qu’ un hybride avec une seule inadéquation puisse pas être stable. La Tm peut être déterminé expérimentalement mais elle est souvent calculée à partir de la formule simple : Tm = (4 × [G + C]) + (2 × [A + T])°C dans laquelle [G + C] est le nombre de nucléotides G et C dans la séquence d’amorce, et [A + T] est le nombre de nucléotides A et T. La température d’hybridation (recuit) pour une expérience de PCR est donc déterminée en calculant La Tm pour chaque amorce et en utilisant une température de 1-2 ° C en dessous de ce chiffre. Remarquez que cela signifie que les deux amorces doivent être conçues de telle sorte qu’elles aient des Tms identiques. Si ce n’est pas le cas, la température d’hybridation( recuit) appropriée pour une amorce peut être trop élevée ou trop faible pour l’autre amorce.
3) La température d’extension, à laquelle se produit la synthèse d’ADN. Il est généralement fixé à 74 ° C, juste en dessous de l’optimum pour la polymerase Taq.
Les produits de PCR:
La PCR est souvent le point de départ d’une série d’expériences dans lesquelles le produit d’amplification est étudié de diverses manières afin d’obtenir des informations sur la molécule d’ADN qui a servi de modèle d’origine. Bien qu’un large éventail de procédures a été conçu pour étudier les produits de PCR, trois techniques sont particulièrement importantes:
l) l’ électrophorèse sur gel des produits de PCR
2) Le clonage des produits de PCR
3) Le séquençage des produits de PCR.
L’ électrophorèse sur gel des produits de PCR :
Les résultats de la plupart des expériences de PCR sont vérifiés en faisant tourner une partie du mélange réactionnel amplifié dans un gel d’agarose. Une bande représentant l’ADN amplifié peut être visible après coloration, ou si le rendement en ADN est faible, le produit peut être détecté par la méthode d’hybridation de Southern. Si la bande attendue est absente, ou si des bandes supplémentaires sont présentes, quelque chose a mal tourné et l’expérience doit être répétée. Dans certains cas, l’électrophorèse sur gel d’agarose est utilisée non seulement pour déterminer si une expérience de PCR a fonctionné, mais aussi pour obtenir des informations supplémentaires. Par exemple, la présence de sites de restriction dans la région amplifiée de l’ADN matrice peut être déterminée en traitant le produit de PCR avec une endonucléase de restriction avant de faire tourner l’échantillon dans le gel d’agarose.
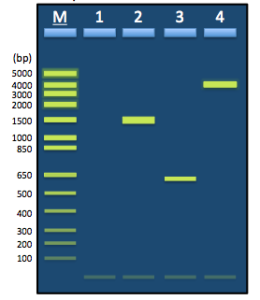
Il s’agit d’un type d’analyse du polymorphisme de la longueur des fragments de restriction (RFLP) et est important à la fois dans la construction des cartes du génome et dans l’étude des maladies génétiques . D’autre part, la taille exacte du produit de PCR peut être utilisée pour établir si l’ADN matrice contient une mutation d’insertion ou de déletion dans la région amplifiée . Les mutations de longueur de ce type forment la base du profilage d’ADN, une technique centrale en médecine légale. Dans certaines expériences, la simple présence ou absence du produit de PCR est la caractéristique diagnostique. Un exemple est lorsque la PCR est utilisée comme procédé de criblage pour identifier un gène désiré à partir d’une banque génomique ou d’ADNc. La réalisation de PCR avec chaque clone dans une bibliothèque génomique peut sembler une tâche fastidieuse, mais un des avantages de la PCR est que les expériences individuelles sont rapides à mettre en place et de nombreuses PCR peuvent être effectuées en parallèle. La charge de travail peut également être réduite par un criblage combinatoire, dont un exemple est illustré à la figure.
Clonage des produits de PCR :
Certaines applications nécessitent qu’après une PCR, les produits résultants soient ligaturés à un vecteur et examiné par l’une quelconque des méthodes standard utilisées pour étudier l’ADN cloné. Cela peut sembler facile, mais il y a des complications.
Le premier problème concerne les extrémités des produits de PCR. D’un examen de Figure ci dessous, on pourrait imaginer que les produits courts résultant de l’amplification par PCR Sont à extrémités franches. Si tel était le cas, ils pourraient être insérés dans un vecteur de clonage par une ligature à extrémités franches ou, en variante, les produits de PCR pourraient être munis d’extrémités collantes Par la fixation d’éléments de liaison ou d’adaptateurs .
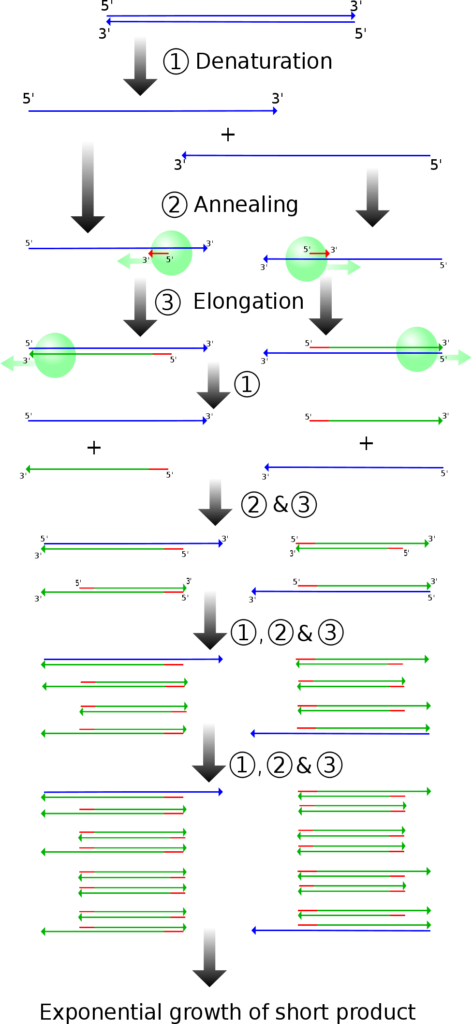
Malheureusement, la situation n’est pas simple. La Taq polymerase tend à ajouter un nucleotide supplémentaire, habituellement une Adénosine, à la fin de chaque brin qu’il synthétise. Cela signifie qu’un produit double-brin de PCR n’est pas à extrémités franches, et la plupart des extrémités 3 ‘ ont une extrémité unique.

Les surplombs pourraient être enlevés par traitement avec une enzyme exonucléase, ce qui donne des produits de PCR avec de vraies extrémités franches, mais ce n’est pas Une approche populaire car il est difficile d’empêcher l’exonucléase de devenir hyperactive et provoquer des dommages supplémentaires aux extrémités des molécules.
Une solution consiste à utiliser un vecteur de clonage spécial qui porte des surplombs de thymidine (T) et qui peut donc être ligaturé à un produit de PCR .

Ces vecteurs sont Habituellement préparé en restreignant un vecteur standard au niveau d’un site à extrémités franches, puis en traitant Avec Taq polymerase en présence de 2′-désoxythymidine 5′-triphosphate (dTTP). Aucune amorce n’est présente ainsi la seule chose que la polymérase puisse faire est d’ajouter un nucléotide T aux extrémités 3 ‘ de la molécule de vecteur à extrémités franches, ce qui a pour résultat le vecteur à queue T dans lequel la PCR Produits peuvent être insérés. Des vecteurs spéciaux de ce type ont également été conçus pour être utilisés avec la méthode de ligation de la topoisomérase, et c’est actuellement La méthode la plus populaire de clonage des produits PCR.
Une seconde solution consiste à concevoir des amorces qui contiennent des sites de restriction. Après la PCR, les produits sont traités par l’endonucléase de restriction, qui coupe chaque molécule à l’intérieur de la séquence d’amorce, laissant des fragments à extrémité collante qui peuvent être ligaturés efficacement dans un vecteur de clonage standard. L’approche ne se limite pas aux cas où les amorces s’étendent sur des sites de restriction qui sont présents dans l’ADN matrice. Au lieu de cela, le site de restriction peut être inclus dans une courte extension à l’extrémité 5′ de chaque amorce. Ces extensions ne peuvent pas s’hybrider à la molécule modèle, Mais ils sont copiés au cours de la PCR, résultant en produits de PCR qui portent des terminaux avec des sites de restrictio
Problèmes avec le taux d’erreur de Taq polymérase :
Toutes les ADN polymérases font des erreurs pendant la synthèse de l’ADN, en introduisant des nucléotides incorrects dans le brin d’ADN croissant. La plupart des polymérases, cependant, sont capable de corriger ces erreurs en inversant l’erreur et en re-synthétisant les séquence. Cette propriété est appelée fonction « relecture » et dépend de la polymérase possédant une activité exonucléase 3′ à 5′. La Taq polymérase manque d’une activité de correction d’épreuves et, par conséquent, est incapable de corriger ses erreurs. Cela signifie que l’ADN synthétisé par la Taq polymérase n’est pas toujours une copie exacte de la molécule modèle. Le taux d’erreur a été estimé à une erreur pour chaque 9000 nucléotides d’ADN qui est synthétisé, ce qui pourrait sembler être presque Insignifiant mais qui se traduit par une erreur dans chaque 300 pb pour les produits PCR obtenu après 30 cycles. C’est parce que la PCR implique des copies de copies, de sorte que les erreurs induites par la polymérase s’accumulent progressivement, les fragments produits à la fin d’une PCR contenant des copies d’erreurs antérieures ainsi que de nouvelles erreurs présenté lors de la dernière ronde de synthèse. Pour de nombreuses applications, ce taux d’erreur élevé ne pose pas de problème. En particulier, le séquençage d’un produit de PCR fournit la séquence correcte du modèle, même si les produits de PCR contiennent les erreurs introduites par la Taq polymérase. C’est parce que les erreurs sont réparties aléatoirement, donc pour chaque molécule qui a une erreur à un nucléotide, il y aura de nombreuses molécules avec la séquence correcte. Dans ce contexte le taux d’erreur est en effet insignifiant. Ce n’est pas le cas si les produits PCR sont clonés. Chaque clone résultant contient plusieurs copies d’un seul fragment amplifié, de sorte que l’ADN cloné n’a pas nécessairement la même séquence que la molécule modèle d’origine utilisée dans la PCR. Cette possibilité confère une incertitude à toutes les expériences réalisées avec les produits clonées de PCR et dicte que, chaque fois que c’est possible, l’ADN amplifié doit être étudié directement au lieu d’être cloné.
La PCR en temps réel permet de quantifier la quantité de matière de départ :
The amount of product that is synthesized during a set number of cycles of a PCR
depends on the number of DNA molecules that are present in the starting mixture
(Table 9.1). If there are only a few DNA molecules at the beginning of the PCR then
relatively little product will be made, but if there are many starting molecules then the
product yield will be higher. This relationship enables PCR to be used to quantify the
number of DNA molecules present in an extract.
Table 9.1
Number of short products synthesized after 25 cycles of PCR with different numbers of starting molecule.
NUMBER OF STARTING MOLECULES NUMBER OF SHORT PRODUCTS
1 4,194,304
2 8,388,608
5 20,971,520
10 41,943,040
25 104,857,600
50 209,715,200
100 419,430,400
Test 1 2 3 4
9.4.1 Carrying out a quantitative PCR experiment
In quantitative PCR (qPCR) the amount of product synthesized during a test PCR is
compared with the amounts synthesized during PCRs with known quantities of starting
DNA. In the early procedures, agarose gel electrophoresis was used to make these
comparisons. After staining the gel, the band intensities were examined to identify the
control PCR whose product was most similar to that of the test (Figure 9.17). Although
easy to perform, this type of qPCR is imprecise, because large differences in the amount
of starting DNA give relatively small differences in the band intensities of the resulting
PCR products.
Today, quantification is carried out by real-time PCR, a modification of the standard
PCR technique in which synthesis of the product is measured over time, as the PCR
proceeds through its series of cycles. There are two ways of following product synthesis
in real time:
l A dye that gives a fluorescent signal when it binds to double-stranded DNA
can be included in the PCR mixture. This method measures the total amount of
double-stranded DNA in the PCR at any particular time, which may over-estimate
the actual amount of the product because sometimes the primers anneal to one
another in various non-specific ways, increasing the amount of double-stranded
DNA that is present.
l A short oligonucleotide called a reporter probe, which gives a fluorescent signal
when it hybridizes to the PCR product, can be used. Because the probe only
hybridizes to the PCR product, this method is less prone to inaccuracies caused by
primer-primer annealing. Each probe molecule has pair of labels. A fluorescent dye
Chapter 10 The Polymerase Chain Reaction 159
Figure 9.17
Using agarose gel electrophoresis to quantify the amount
of DNA in a test PCR. Lanes 1 to 4 are control PCRs
carried out with decreasing amounts of template DNA.
The intensity of staining for the test band suggests that
this PCR contained approximately the same amount of
DNA as the control run in lane 2.
Note: The numbers assume that amplification is 100% efficient, none of the reactants becoming limiting during the course of the PCR.
Quenching
compound
Fluorescent
label
Fluorescent
signal
Probe
Oligonucleotide
probe
DNA target
DNA
Figure 9.18
Hybridization of a reporter probe to its target DNA.
Amount of PCR product
Number of cycles
Threshold
Figure 9.19
Quantification by real-time PCR. The graph shows
product synthesis during three PCRs, each with a
different amount of starting DNA. During a PCR, product
accumulates exponentially, the amount present at any
particular cycle proportional to the amount of starting
DNA. The blue curve is therefore the PCR with the
greatest amount of starting DNA, and the green curve is
the one with the least starting DNA. If the amounts of
starting DNA in these three PCRs are known, then the
amount in a test PCR can be quantified by comparison
with these controls. In practice, the comparison is made
by identifying the cycle at which product synthesis
moves above a threshold amount, indicated by the
horizontal line on the graph.
is attached to one end of the oligonucleotide, and a quenching compound, which
inhibits the fluorescent signal, is attached to the other end (Figure 9.18). Normally
there is no fluorescence because the oligonucleotide is designed in such a way that
its two ends base pair to one another, placing the quencher next to the dye.
Hybridization between the oligonucleotide and the PCR product disrupts this base
pairing, moving the quencher away from the dye and enabling the fluorescent
signal to be generated.
Both systems enable synthesis of the PCR product to be followed by measuring the
fluorescent signal. Quantification again requires comparison between test and control
PCRs, usually by identifying the stage in the PCR at which the amount of fluorescent
signal reaches a pre-set threshold (Figure 9.19). The more rapidly the threshold is
reached, the greater the amount of DNA in the starting mixture.
9.4.2 Real-time PCR can also quantify RNA
Real-time PCR is often used to quantify the amount of DNA in an extract, for example
to follow the progression of a viral infection by measuring the amount of pathogen DNA
that is present in a tissue. More frequently, however, the method is used as a means of
measuring RNA amounts, in particular to determine the extent of expression of a
particular gene by quantifying its mRNA. The gene under study might be one that is
switched on in cancerous cells, in which case quantifying its mRNA will enable the
160 Part I The Basic Principles of Gene Cloning and DNA Analysis
Standard PCR