Des procédés de séparation :
Un des devoirs d’un chimiste est d’analyser des échantillons de produits chimiques. Pour déterminer la composition de l’échantillon, une séparation des différents éléments est nécessaire pour éviter les interférences entre les espèces.
Il peut être important de déterminer quels éléments chimiques sont présents, dans quelles quantités et dans quel état d’oxydation. Plusieurs méthodes de séparation peuvent satisfaire les besoins de la pharmacie pour obtenir une bonne séparation. Dans la plupart des cas l’échantillon doit être une solution dont le solvant ne contient pas d’espèces interférentes.
Les méthodes de séparation sont basées sur l’affinité des espèces, pour différents solvants, sur les processus d’équilibre et sur la cinétique. Certaines méthodes permettent d’obtenir l’échantillon de retour et certaines le détruisent.
Extraction :
Cette méthode est basée sur l’affinité de l’espèce avec deux phases liquides non miscibles différents. Une phase est aqueuse et la seconde est organique. L’expérience est réalisée dans une ampoule à décanter (ou entonnoir de séparation). C’est un morceau de verre ou de matière plastique en forme de cône renversé avec une vanne sur le fond et une ouverture à son sommet. Un bouchon peut fermer l’entonnoir pour permettre un mélange énergique.

L’échantillon est préparé dans une des deux phases et est placé dans l’entonnoir d’extraction par son ouverture supérieure. La deuxième phase est ajoutée (l’ordre d’insertion des deux phases n’a pas vraiment d’importance). Comme les phases ne sont pas miscibles, les échanges de particules entre les phases ne peuvent être faites à l’interface et des espèces chargées ne se déplacent pas dans la phase organique : leur solubilité dans toute phase organique est très faible. Pour améliorer les échanges les liquides sont mélangés en secouant l’entonnoir et en tournant à l’envers. Cela augmente la surface de contact entre les deux phases ainsi l’échange est amélioré. Au cours de la phase de mélange il est recommandé d’ouvrir brièvement l’entonnoir (par le capuchon ou par la valve) afin de libérer un éventuel excédent de pression. Rappelez-vous quelques notions de sécurité : il peut être nécessaire d’ouvrir l’entonnoir sous une hotte et ne pas mettre votre nez ou un oeil juste au-dessus de l’ouverture lorsque vous relâchez la pression. Ensuite le système est mis au repos jusqu’à ce que l’interface soit mince et claire. La phase de densité la plus faible se trouve en haut et la plus dense est au fond. Habituellement la phase organique au-dessus de la phase aqueuse en dessous car la densité des solvants organiques est généralement inférieure à la densité de l’eau (huile flotte sur l’eau). On peut ainsi éliminer la phase aqueuse hors de l’entonnoir par l’ouverture de la valve au fond de la fiole.
Répartition des espèces :
A l’équilibre chaque espèce est répartie entre les deux phases. On peut définir un coefficient de distribution KD qui est le rapport entre la concentration d’une espèce dans la phase organique et dans la phase aqueuse :

Si nous ne considérons pas spécifiquement l’équilibre c’est le rapport de distribution D.
![]()
Pour les concentrations de l’espèce nous considérons toutes les formes d’une espèce donnée. Certaines réactions peuvent se produire dans une phase mais pas dans l’autre et déplacer le rapport de distribution. Par exemple un acide dans la phase aqueuse peut être sous sa forme acide HA ou sous forme de base conjuguée A- mais la solubilité des ions dans les phases organiques est très faible.
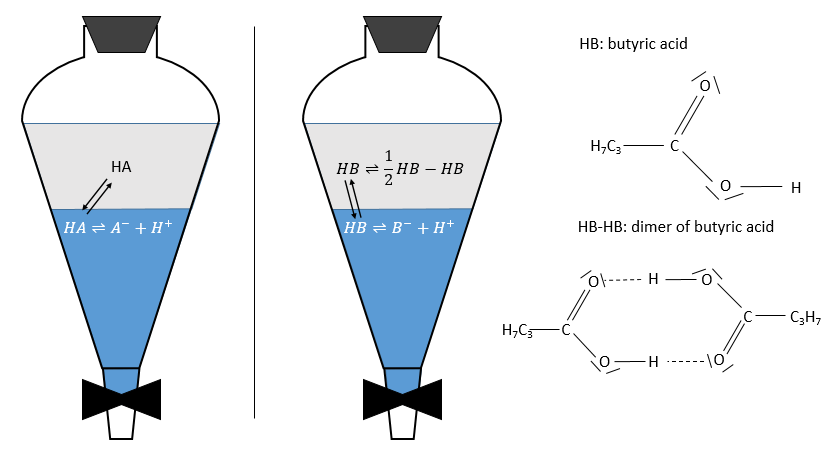
En conséquence les A- ne sont pas échangées et restent dans la phase aqueuse. D’autre part les espèces neutres HA ont une concentration équilibrée entre la phase aqueuse et la phase organique. Ainsi le rapport de distribution de l’acide est :
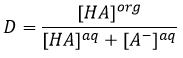
En conséquence le rapport de distribution peut dépendre de l’acidité de la phase aqueuse. Des équilibres peuvent également se produire dans la phase organique comme il est montré par l’exemple de l’acide butyrique. Dans la phase organique cet acide carboxylique peut former un dimère avec deux liaisons hydrogène. Dans ce cas le rapport de distribution sera
Le choix des solvants est donc crucial pour une bonne extraction. Les solvants peuvent être polaire ou non polaire avec la restriction qu’ils ne doivent pas être miscible.
Extraction des espèces organiques :
Pour extraire un composé organique de la phase organique, nous devons obtenir des espèces chargées. Les espèces neutres sont partagées entre les deux phases mais les ions restent dans la phase aqueuse. Si le composé n’est pas ionisable nous ne pouvons jouer que sur la nature du solvant pour avoir la meilleure extraction, à savoir lorsque KD est petit. Si le composé peut être dissocié on peut régler l’acidité de la solution aqueuse afin d’optimiser l’extraction.
L’acide acétique a un pKa de 4.7
![]()
A ce pH, dans la solution aqueuse, il y aura autant de CH3COOH que de CH3COO–. L’acétate peut être extrait dans la phase aqueuse. Si l’on utilise un acide plus fort la majorité de l’acide acétique qui pénètre dans la solution aqueuse va se dissocier pour former de l’acétate qui peut être extraite.
Un autre cas est le cas des espèces amphotères. Ces espèces peuvent jouer le rôle d’un donneur ou d’un accepteur de protons. L’oxime (8-hydroxyquinoléine) est un amphotère.

A pH <5 l’azote est protoné, donnant une charge positive au composé qui peut maintenant être extrait. A un pH> 10 le groupe hydroxyle perd son proton ce qui conduit à une espèce chargée négativement qui peut également être extraite.
Entre 5 <pH <10 oxime reste dans la phase organique.
L’extraction de métaux :
Les cations métalliques peuvent être extraits à partir de solutions aqueuses. Dans ce cas nous voulons obtenir une grande valeur de D. Pour transférer les métaux de la phase aqueuse leur charge doit être cachée par la formation de paires ioniques fortes ou par la formation de complexes de coordination. L’ oxime peut former un complexe avec des métaux pour les stabiliser dans la phase organique. Par exemple si l’on veut extraire l’aluminium d’une solution aqueuse, l’oxime peut se déplacer à partir de la phase organique à la phase aqueuse. Dans de bonnes conditions de pH, l’oxime perd son proton. Dans sa forme de base l’oxime forme un complexe avec les ions d’aluminium, un complexe qui peut maintenant être transféré dans la phase organique.
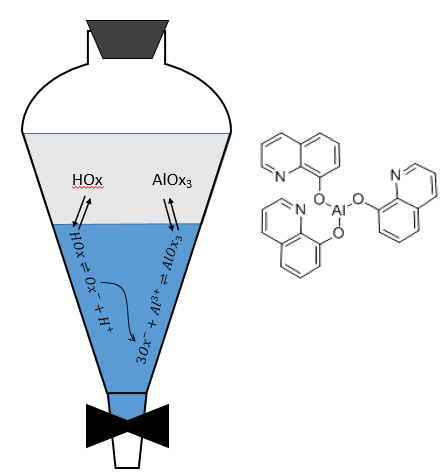
Fe3 + peut former une paire ionique dans une solution concentrée de HCl. La paire ionique (FeCl4) -H3O + peut se déplacer dans la phase organique. Notez que ce produit de la réaction se déplace hors de la solution aqueuse. L’équilibre est donc déplacé vers la droite.
Une autre façon de déplacer l’équilibre vers la droite est d’augmenter le pH de la solution aqueuse. Il est possible d’extraire plusieurs métaux spécifiquement en fonction du pH de la solution aqueuse. La figure suivante représente l’extraction de différents métaux par formation de dithizomates en fonction du pH.
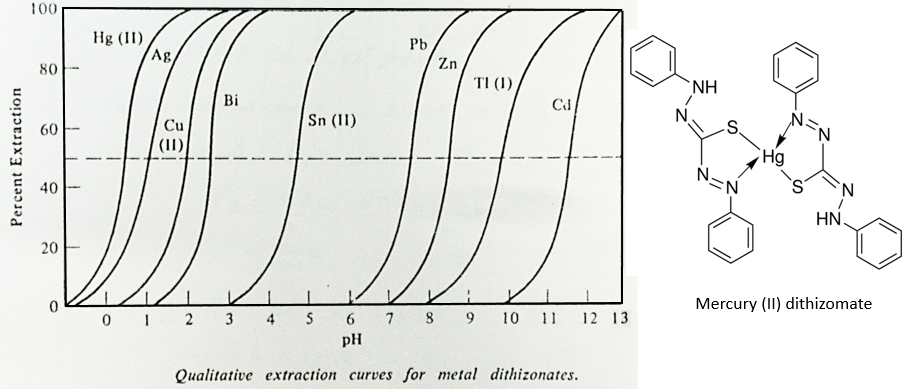
On peut voir que l’extraction du plomb commence à pH> 6 tandis que l’Ag est déjà entièrement extrait à pH = 3. Pour la séparation du Cu (II) de l’Ag il faudra un autre procédé de séparation.
Fraction extraite :
Prenons l’extraction de l’acide butyrique. Le rapport de distribution D est le rapport de sa concentration dans la phase organique (éther) par rapport à sa concentration dans la phase aqueuse (eau). Après l’extraction 3g d’acide butyrique est dans la phase organique et 1g est encore dans la phase aqueuse.
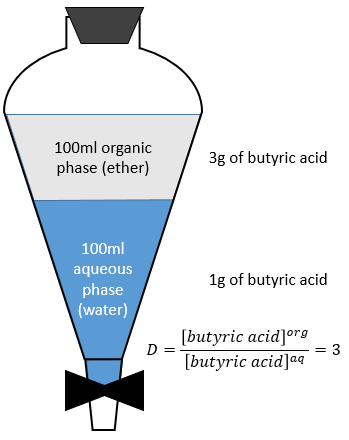
La fraction extraite F est la quantité de l’acide dans la phase organique sur la quantité totale de l’acide :

F = 3/4 si les volumes des phases aqueuses et organiques sont égales. Le rapport des volumes RV est le volume de la phase organique sur celui de la phase aqueuse. Si l’on considère un grand volume de solvant, à l’égard du volume de l’espèce à séparer, les volumes initiaux de solvants mis en présence peuvent être utilisés avec une bonne approximation. Après l’injection RV dans l’expression de F on obtient l’expression suivante :
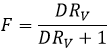
Nous pouvons conclure que l’extraction a de meilleurs résultats lorsque D ou RV est grande.
La fraction restante G est la fraction de l’échantillon qui n’a pas été transféré à la phase organique et est simplement
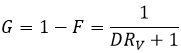
Maintenant si deux composés A et B doivent être séparés, nous devons concevoir l’expérience de telle manière que la multiplication de DA par DB soit égale à 1. En effet, si DA et DB sont grands mais DA >>> DB, la séparation ne sera pas efficace. Imaginez que DA = 1000 et DB = 20. (A) a une meilleure extraction que B mais les deux espèces sont extraites : les fractions extraites( en envisagent des volumes identiques de phases) seront :
![]()
Malgré l’énorme différence de D la séparation est mauvaise. Maintenant si DA = 10 et DB = 0,1 l’extraction de ces deux composés est plus faible que dans l’exemple précédent mais la séparation est bien meilleure :
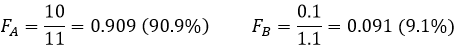
Cette fois-ci 91% de A sont dans la phase organique pour seulement 9% de B.
Pour obtenir une meilleure séparation nous pouvons répéter l’extraction à partir de la phase organique que nous venons d’extraire. Revenons à notre premier exemple de l’acide butyrique. Pour ce composé nous avions trouvé D = 3. Nous pouvons jouer sur les volumes de la phase pour modifier l’efficacité de l’extraction. Si nous mettons la moitié du volume de l’éther puis RV = 0,5 la fraction extraite sera :
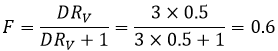
Sur les 4 premiers grammes 1,6 g sont dans la phase aqueuse et 2,4 g sont dans la phase organique. On pourrait avoir une meilleure extraction avec un plus grand volume d’éther. Cependant, si nous faisons une seconde extraction avec les mêmes conditions, F est à nouveau 0,6 et nous aurons 0,64 g d’acide butyrique restant dans la phase aqueuse.
Nous avons fait deux extractions avec 50 ml d’éther à la place d’une extraction avec 100 ml d’éther. Donc, avec le même volume, mais avec deux extractions, on a extrait plus de l’acide butyrique (3,36 g de 3 g vs). Plusieurs extractions avec un petit volume sont donc plus efficaces qu’ une extraction avec un volume important. Après n extraction, la fraction restante est

Appareil de Craig
Ce procédé a été optimisé dans l’appareil de Craig. Des dizaines à des centaines de tubes en forme de H sont connectés. La figure suivante montre un tube:
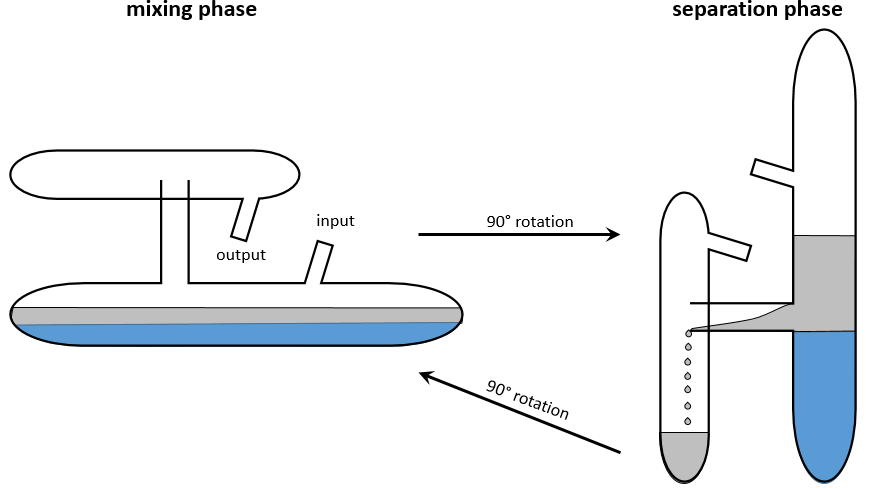
Les bras mécaniques mélangent les petits tubes au cours de la phase de mélange et les inclinent pour transférer la phase supérieure dans le petit compartiment du tube en forme de H. Une fois le transfert effectué les tubes sont remis dans leur position initiale. Cette rotation provoque la vidange du petit compartiment dans le tube suivant. Le processus est répété dans tous les tubes simultanément.
Dans un premier temps l’échantillon se trouve dans le premier tube (noté 0) et la phase inférieure est déjà dans tous les tubes :
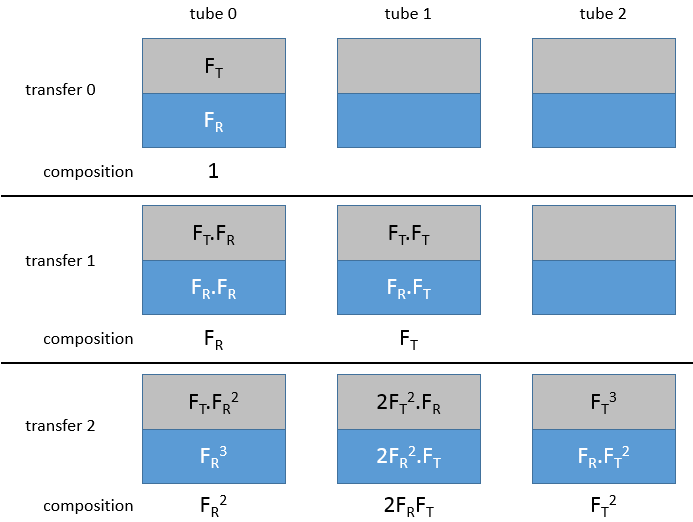
Au début tout le substrat est dans le tube 0, la moitié de celui-ci est dans la phase organique et l’autre moitié dans la phase aqueuse. La phase organique est transférée dans le tube 1. On peut définir une fraction FR restant qui est la fraction de l’échantillon qui reste dans le tube lors d’un transfert et une fraction de transfert FT qui est la fraction qui est transférée dans le tube suivant .. Après le premier transfert il y a FR dans t0 et FT en t1. Le tube 0 est maintenant rempli avec le solvant et les tubes sont mélangés.
Après avoir mélangé le tube 0 la fraction restante FR sera séparée en 2 phases : organique et aqueuse. Dans la phase aqueuse (en considérant que la phase organique est transférée à chaque fois) il restera la fraction restante de la première fraction restante à savoir FR.FR. Dans la phase organique, il y aura la fraction transférée de la fraction restante à savoir FR.FT.
Dans le tube 1 la fraction transférée à partir du tube 0 est séparée entre les deux phases. Dans la phase aqueuse il sera maintenant FR.FT et dans la phase organique FT.FT. Une fois que le mélange est effectué, les phases organiques sont transférées sur le tube suivant. Les phases aqueuses restent dans leur tube. FT2 est transféré de t1 à t2 et FR.FT est transféré de t0 à t1. Après le transfert on trouve respectivement dans les tubes 0, 1 et 2, FR2, 2FR.FT and FT2.
Après trois transferts nous aurions FR3, 3FR2.FT, 3FR.FT2 and 3FR3. Vous avez peut-être remarqué que ces valeurs sont les membres d’un exposant d’une somme :
![]()
n étant le nombre de transfert. Dans un tube r donné la fraction est :
![]()
On peut donc prédire la composition de chaque tube en connaissant le résultat d’une seule extraction. Pour un grand nombre d’extractions nous trouvons l’équation d’une courbe de Gauss.
La largeur du pic est caractérisée par un écart-type :
![]()
Le pic se déplace linéairement avec n répartis comme √n et l’intensité des baisses de pointe en 1 / √n.
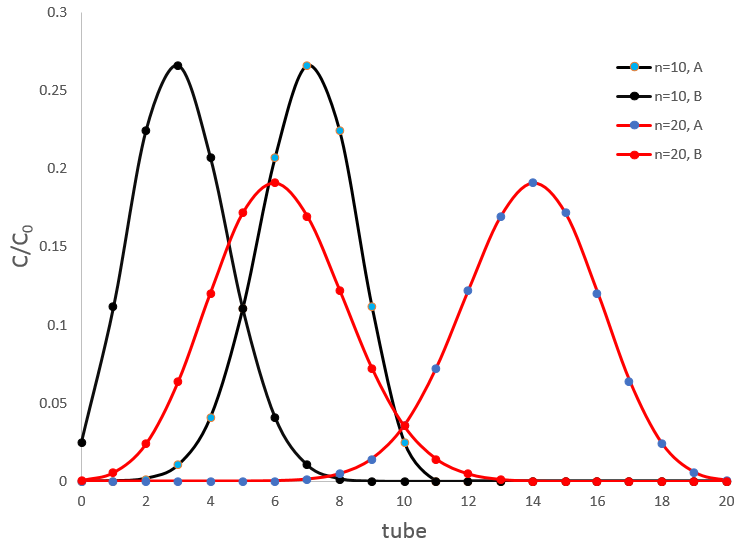
Si deux substrats sont dans l’échantillon l’appareil de Craig va les séparer. Encore une fois nous avons à choisir attentivement les conditions de l’extraction pour obtenir une bonne séparation. La figure ci-dessus montre les concentrations de deux espèces avec DA=1/DB=9/4 après 10 et 20 transferts. Il y a un chevauchement entre les courbes où les espèces ne sont pas bien séparées mais il diminue considérablement avec le nombre d’extractions. Au maximum du pic pour A, après 10 transferts, nous trouvons ~ 27% de la concentration initiale de A mais également 1% de la concentration initiale de B. Après 20 transferts on a 19% de [A]0 et seulement 0,03% de [B]0.
Les avantages de l’appareil de Craig sont que tout est mécanique de sorte que la précision est bonne et que l’on peut obtenir soit le résultat de l’extraction à la fin de l’appareil ou à un tube donné dans l’appareil.