L’intérêt des ajouts (additions) électrophiles sur les liaisons C = C est de transformer deux carbones sp2 en deux carbones sp3 et d’ajouter une chaîne ou un groupe sur la molécule existante.
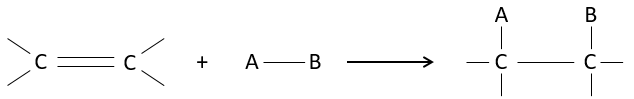
Comme deux atomes de carbone sont impliqués dans la réaction, l’ajout du nouveau groupe peut donner plusieurs produits avec plus ou moins de (stéréo) sélectivité. En outre la transformation en carbones sp3 introduit également une régiosélectivité qui doit souvent être prise en considération.
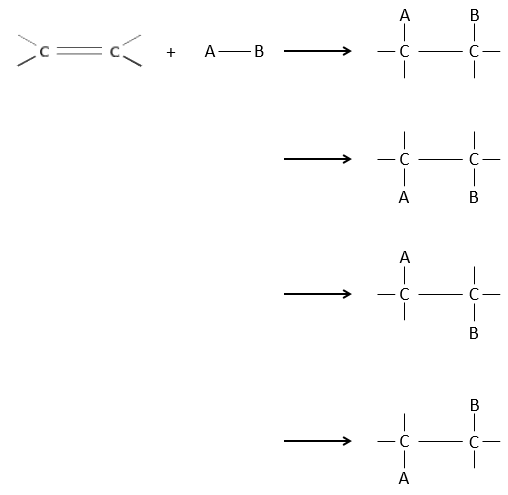
Par conséquent nous allons discuter dans cette section les méthodes et les règles qui doivent être suivies pour obtenir le produit souhaité au cours de l’addition d’un groupe électrophile sur les liaisons doubles ou triples.
L’hydrogénation catalytique :
Il est, en principe, l’ajout (l’addition) le plus facile sur une liaison C = C car il n’y a pas de nouveau groupe sur le produit. Les carbones sp2 sont réduits en carbones sp3. Or cette réaction n’est pas spontanée en dépit du fait que c’est une réaction exothermique. L’énergie d’activation pour briser la liaison H-H est énorme et un catalyseur est nécessaire pour effectuer la réaction.
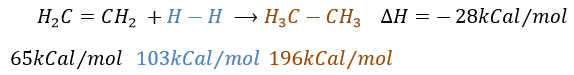
Comme catalyseur on peut utiliser le platine (Pt), le nickel de Raney(Ni) et du palladium sur C (Pd/C).
Ils ont la capacité de dissocier les atomes d’hydrogène et de les fixer sur leur surface ce qui les rend disponibles pour l’ajout sur les alcènes. Comme nous avons vu dans la section de la cinétique, le catalyseur diminue l’énergie d’activation de la réaction et le processus peut être représenté en 4 étapes :
– Approche et la liaison des réactifs
– Déplacement sur la surface
– Réaction(s)
– Départ des produits
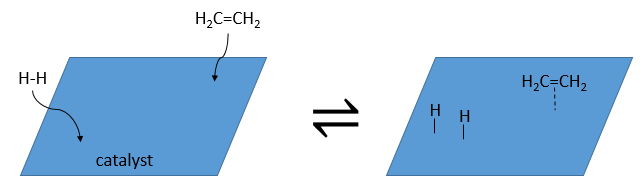
Lors de la première étape les réactifs ont lieu sur la surface du catalyseur hétérogène. La quantité de réactif qui peut avoir lieu sur le catalyseur dépend de sa surface. Il est donc conseillé d’utiliser de petites particules de catalyseur pour augmenter le rapport surface/volume (ou surface/masse). Les alcènes se placent parallèlement à la surface dans la direction de la liaison π.
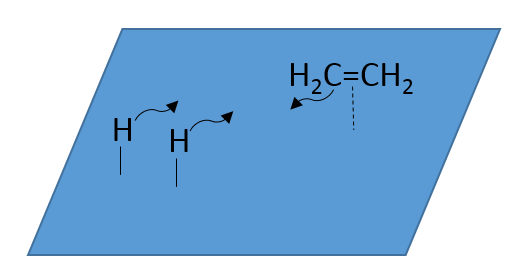
Il n’y a aucune raison que les réactifs se lient immédiatement sur des spots à proximité. La deuxième étape consiste donc à ce que les réactifs se déplacent sur la surface. Ils finiront par être en contact les uns avec les autres de sorte que la réaction peut avoir lieu.
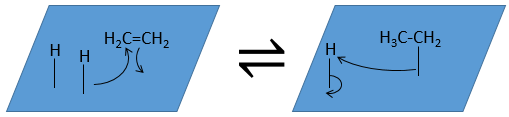
La troisième étape (et éventuellement les suivantes) est la réaction entre les réactifs. Un hydrogène activé attaque un carbone sp2 tandis que la liaison π relie le second carbone avec la platine. La prochaine étape de la réaction est que la liaison entre le carbone et le platine attaque l’hydrogène restant.
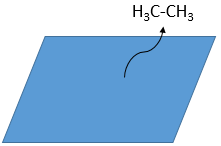
Durant la dernière étape, l’alcane quitte la surface du catalyseur en laissant un espace pour que les réactifs se lient.
L’hydrogénation est stéréosélective, à savoir qu’au niveau de la liaison C = C la place des groupes sur les atomes de carbone ne sont pas équivalents et la molécule est plane. Si les atomes d’hydrogène sont fixés sur le même côté (mais pas sur le même atome de carbone) de la molécule alors c’est une addition syn. Comme les groupes ajoutés sont des hydrogènes , le produit est donc cis (voir substitutions nucléophiles et cis et trans des produits).
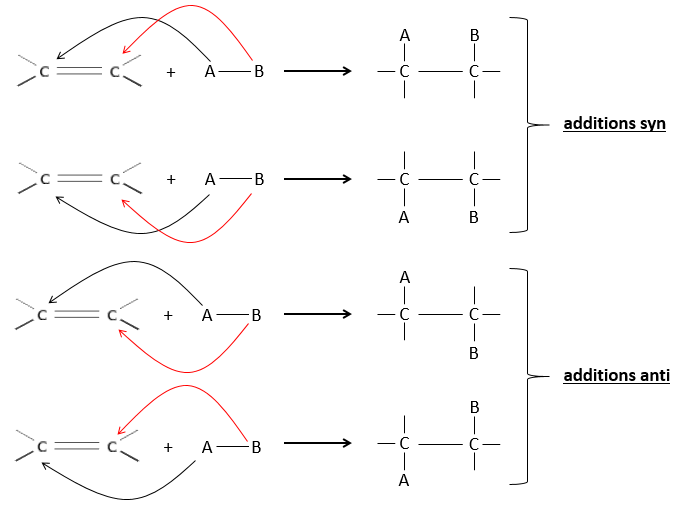
Si les groupes ont été ajoutés sur les côtés opposés des molécules (toujours pas sur le même carbone) l’addition serait anti et le produit serait trans. Ce produit ne sera pas observé parce que les réactifs sont liés au catalyseur et ne peuvent pas se déplacer librement pendant la réaction. Les deux hydrogènes sont ainsi du même côté de la molécule avant la réaction et après la réaction.
Ceci est important seulement si, après la réaction, le carbone sp3 nouvellement formé ne peut pas tourner. Ce sera le cas lorsque la liaison est impliquée dans un cycle ou s’ il y a des groupes volumineux. En outre le fait que l’on obtient seulement le produit cis ne signifie pas qu’un seul produit est possible. La raison en est qu’il y a un seul plan de chaque côté de la liaison de π (pour un total de 2). Si les deux plans sont équivalents, nous obtenons un mélange racémique 50-50 de deux produits cis.
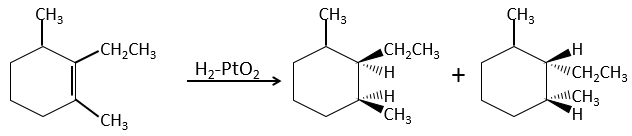
Si les plans ne sont pas équivalents un des produits est favorisé : les atomes d’hydrogène sont fixés sur le côté moins occupé parce que les alcènes se placent sur le catalyseur de sorte que les groupes volumineux soient loin du catalyseur.
Les catalyseurs solubles peuvent également être utilisés pour cette réaction. C’est le cas pour le catalyseur de Wilkinson (tris(triphénylphosphine) rhodium (I).
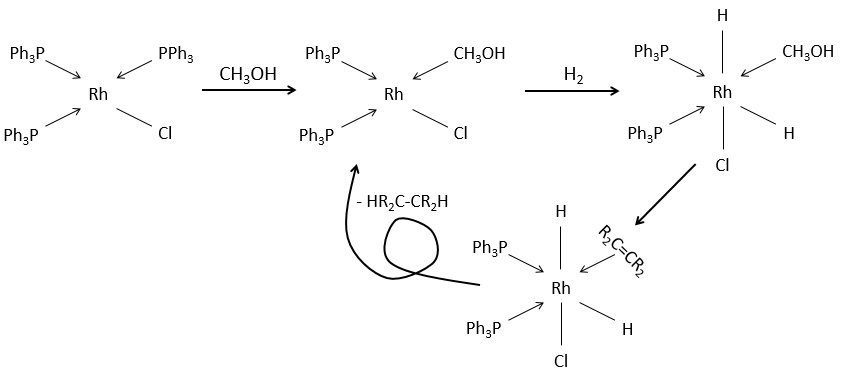
Il conduit également aux produits cis.
En termes de réactivité les atomes de carbone substitués réagissent plus lentement que les carbones ayant moins de groupes en raison de l’encombrement stérique : les atomes d’hydrogène ne sont pas très mobiles sur la surface du catalyseur et la liaison π doit être proche pour obtenir l’hydrogénation.
Les alcynes réagissent aussi plus rapidement que les alcènes ce qui permet d’hydrogéner tous les alcynes en alcènes avant la formation des alcanes. Nous pouvons ainsi limiter l’hydrogénation des alcènes si nous utilisons un mauvais catalyseur et si nous mettons un équivalent de H2 par alcyne. Nous allons obtenir l’alcane avec 2 équivalents de H2 par alcyne.
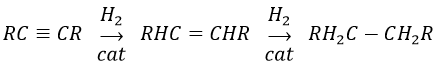
Encore une fois cela conduira à l’alcène cis. Pour obtenir l’alcène trans nous utilisons du sodium dans l’ammoniac. Le sodium génère un radical sur un atome de carbone et une charge négative sur l’autre.
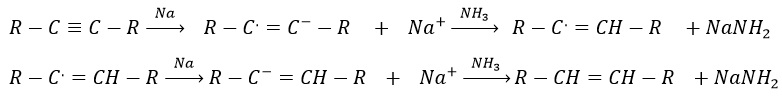
Le produit trans est favorisé pour des raisons stériques.
Les produits aromatiques peuvent difficilement être hydrogénés avec H2/cat (à 150° C), mais ils peuvent également être réduits différemment par la réduction de Birch impliquant Na, NH 3 et un alcool.
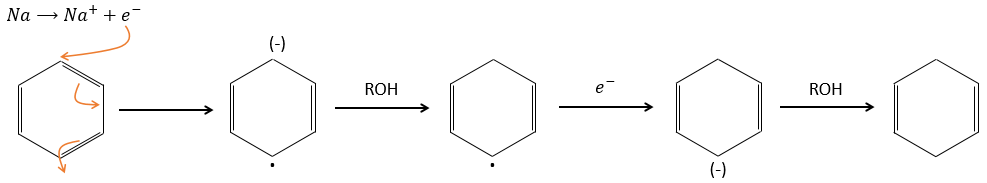
Autres types d’ajouts électrophiles :
L’addition de HX :
Les autres réactifs HX contenant un atome d’hydrogène peuvent être plus facilement cassés (ou sont déjà ioniques) et ne nécessitent pas de catalyseur. Dans le cas d’un acide halogène, en raison de leur caractère nucléophile, les liaisons de π qui sont plus faibles que les liaisons σ ont tendance à attaquer les molécules électrophiles. Dans ce cas la cible électrophile est le proton. Il est appelé une attaque électrophile. Le X nucléophile attaque le carbone positif (ou carbocation) par la suite.
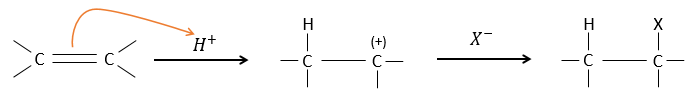
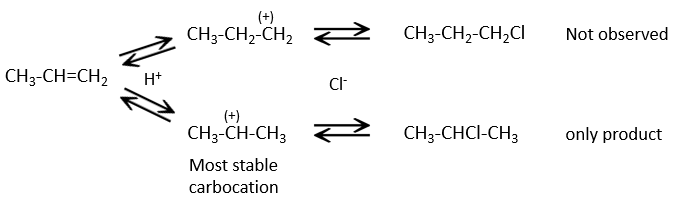
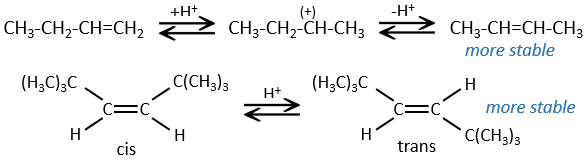
Cette réaction ne comporte pas de stéréosélectivité parce qu’il y a deux étapes distinctes mais implique la régiosélectivité : c’est la règle de Markovnikov qui dit que l’halogène est fixé sur le carbone le plus substitué ou, inversement que le proton, est fixé sur le carbone le moins substitué. Personnellement je préfère rappeler que le carbocation le plus stable est formé. Rappelez-vous que les carbocations peuvent réorganiser la molécule pour former le carbocation plus stable.
L’addition d’eau :
L’eau est ajoutée à alcènes en présence d’acide sulfurique :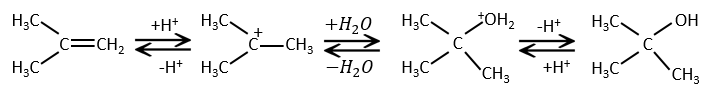
L’acide sulfurique est choisi parce que le sulfate est moins nucléophile que l’eau (parce que la charge est stabilisé par résonance). HCl doit être évité car Cl– est plus nucléophile que H2O. En outre toutes les étapes sont réversibles. Cela signifie que l’alcool nouvellement généré peut être déshydraté dans le cas d’un excès d’acide.
La présence d’un carbocation explique quelques réarrangements d’alcènes qui peuvent être observés dans des solvants acides.
L’addition de X2 :
Des halogènes sont additionnés à C = C pour obtenir des produits trans.
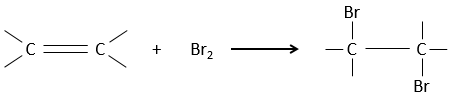
Il est étrange que des éléments aussi chargés en électrons que des halogènes soient utilisés comme électrophiles pour cette réaction. La raison en est que la liaison entre les halogènes est polarisable. Un halogène va donc ressentir les électrons de la liaison π et va transférer une partie de ses électrons à l’autre halogène. L’halogène partiellement positif peut donc être attaqué par la liaison π et forme un halonium intermédiaire qui sera attaqué par l’halogène négatif sur l’autre côté de la liaison C-C.
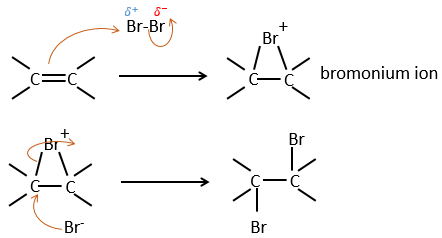
Cette réaction est stéréosélective (toujours trans) et stéréospécifique (le résultat dépend de la disposition du réactif). En présence d’autres agents nucléophiles, tels que l’eau, la réaction commence normalement mais il existe une compétition entre X– et le nucléophile.
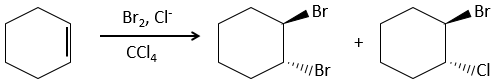
Cette réaction est plus lente sur alcynes parce que le halonium intermédiaire est moins stable. L’halogénation est donc difficile à contrôler : les alcènes réagissent plus vite que les alcynes.
La réaction d’oxymercuration :
Cette réaction est un moyen utile pour former des alcools ou des éthers à partir d’alcènes. Elle implique l’acétate de mercure et de l’eau/alcool dans le THF comme solvant. L’ouverture nucléophile de l’eau/alcool se fait dans la position anti et sur le carbone le plus substitué (il n’y a donc plus d’espace pour l’acétate de mercure).
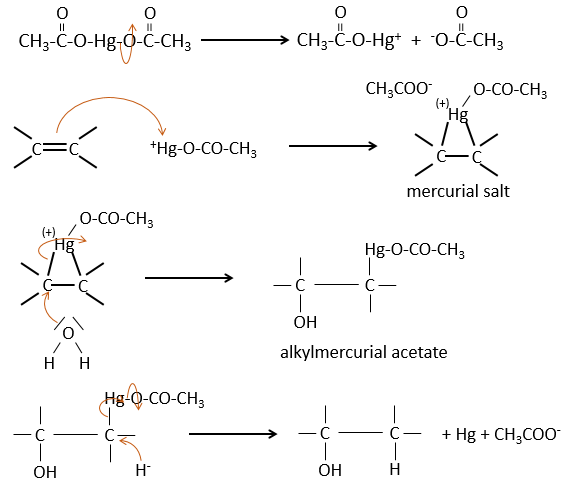
L’oxymercuration suit la règle de Markovnikov mais il n’y a pas de carbocation impliqué dans le processus. Comme tous les ajouts il est régiosélectif.
L’eau conduit à la formation d’un alcool et on peut utiliser un alcool au lieu de l’eau pour obtenir un éther.
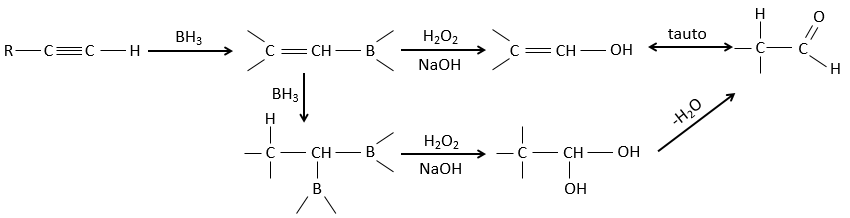
Réaction d’hydroboration :
BH3 (borane) est un acide de Lewis qui est stable sous la forme d’un dimère et peut remplacer son hydrogène en 3 par 3 chaînes de carbone de THF (tétrahydrofurane (CH2)4O). Une fois que les chaînes sont liées, il est possible de supprimer le bore par une oxydation avec H2O2, NaOH et H2O2 pour obtenir le plus grand nombre d’alcools.
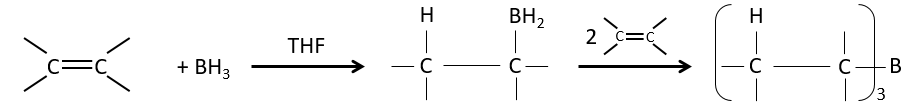
Cette réaction ne suit pas la règle de Markovnikov (anti-Markovnikov) et est stéréosélective. Le mécanisme de la réaction implique l’orbital p vide du bore qui interagit avec la liaison π pour former un complexe. Ce complexe permet un état de transition, avec 4 centres, dans lequel les liaisons se déplacent.
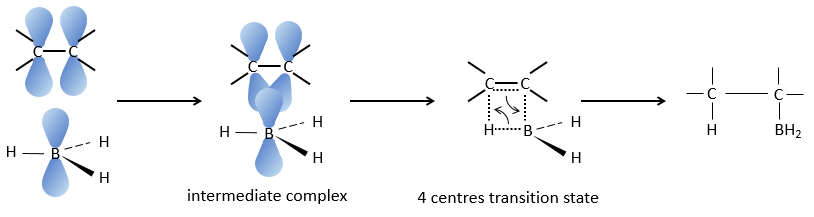
La règle Markovnikov s’applique lorsque l’hydrogène est chargé positivement. Dans le cas présent nous ajoutons H– et BH2+.
L’élimination du bore est fait avec rétention de configuration et est stéréospécifique. Le bore est moins électronégatif que le carbone et donne ainsi ses électrons. NaOH prend un proton du H202 pour former HOO– qui attaque le bore, transferant sa charge négative. Il existe un réarrangement qui met un atome d’oxygène entre la chaîne et le bore.
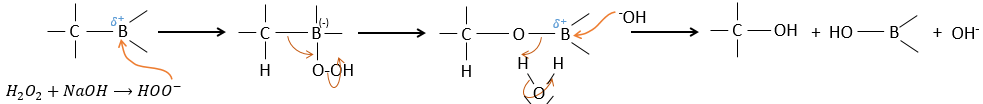
L’eau et OH– agissent à l’étape suivante pour supprimer le bore.
Cette addition peut également se faire sur des alcynes pour former des aldéhydes par tautomérisation.
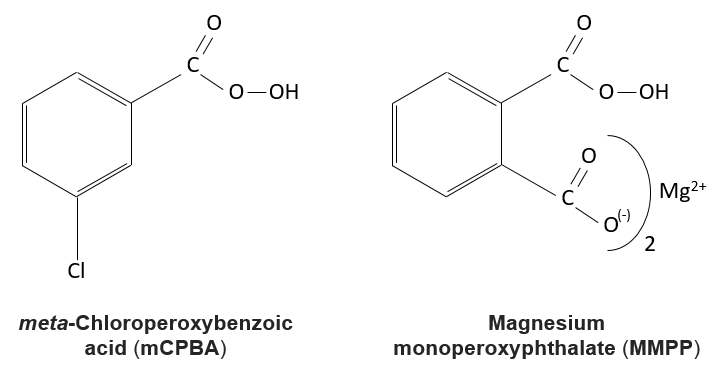
La réaction des alcènes avec des acides peroxycarboxyliques
Les acides peroxycarboxyliques sont généralement instables. Le MCPBA et MMPP sont toutefois des peroxyacides stables et sont largement utilisés dans les laboratoires :
Un des oxygènes des acides peroxycarboxyliques est électrophile. Ces acides peuvent donc réagir avec des alcènes pour former des époxydes et de l’acide carboxylique.
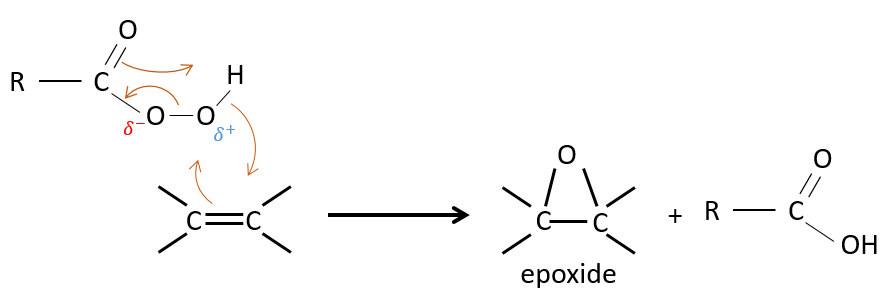
L’addition est donc une réaction concertée, est stéréospécifique et est syn. La réaction est faite dans le plan le moins encombré de l’alcène. La réactivité augmente avec la quantité de substituants qui partagent leurs électrons avec la liaison π (les chaînes alkyles par exemple).
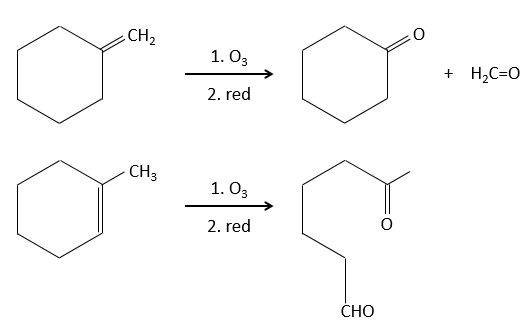
L’ozonolyse :
L’ozone peut briser des liaisons doubles. Dans un premier temps il ouvre la double liaison pour donner un intermédiaire appelé molozonide.

Il rompt pour former l’ozonide qui peut être la prochaine oxydé ou réduit.
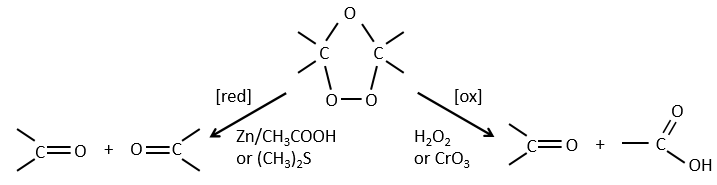
L’ozonolyse est donc un bon moyen pour obtenir les cétones et les acides carboxyliques à partir d’alcènes. La méthode est également utile pour déterminer la position d’une double liaison dans une molécule quand nous ne pouvons pas facilement la déterminer avec les techniques habituelles.