Chapitre 10 : Les réactions d’acides carboxyliques et leurs dérivés
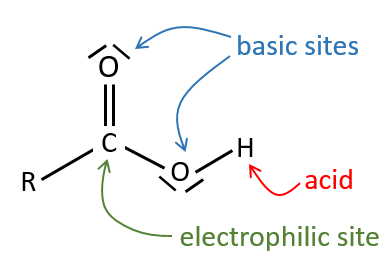
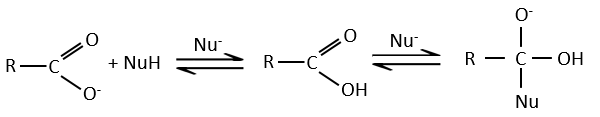
Tous les atomes de groupes acides carboxyliques ont un caractère spécifique. Le groupe est donc ambident: il possède deux ou plusieurs centres réactifs distinguables, alternatifs et en forte interaction. Il a des propriétés acides et basiques.
All the atoms of carboxylic acids groups have a specific character. The group is thus ambident: it possesses two or more alternative and strongly interacting distinguishable reactive centres. It has acidic and basic properties.
Un acide carboxylique est plus acide que l’alcool correspondant car la base conjugué le carboxylate, est stabilisée par résonance (pKa≈5 vs pKa≈17).
L’oxygène du carbonyle est plus basique que l’autre parce que la charge positive est plus stabilisée.
Synthèse d’acides carboxyliques :
Il existe plusieurs façons pour former des acides carboxyliques :
1) par oxydation :
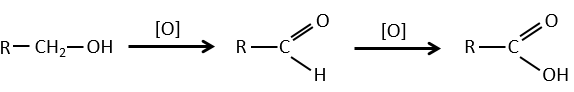
Les acides carboxyliques peuvent être faits par l’oxydation d’aldéhydes ou d’alcools. Nous pouvons utiliser CrO3 ou KMnO4 comme oxydants par exemple.
2) par Grignard :
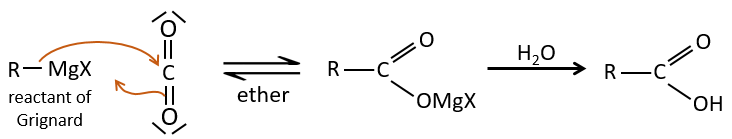
Une réaction de Grignard peut réagir avec du CO2, puis avec de l’eau pour former un acide carboxylique
3) à partir de nitriles :

Dans les environnements acides les nitriles réagissent avec l’eau pour générer de nouveaux acides et libérer une molécule d’ammoniac.

Cette méthode peut être utilisée pour étendre une chaîne carbonique. Une fois qu’un acide carboxylique est obtenu on le réduit pour obtenir un alcool. Le groupe -OH est éliminé par substitution nucléophile et la boucle redémarre. Chaque boucle ajoute un carbone dans la chaîne.
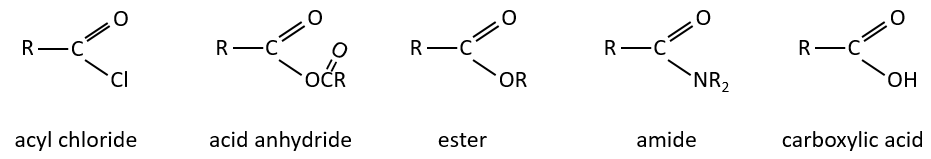
Les dérivés d’acides carboxyliques sont :
Réaction d’addition-élimination :
Les acides carboxyliques et leurs dérivés sont attaqués par des nucléophiles sur le carbone, comme cela est fait pour les cétones et les aldéhydes. Toutefois cet ajout est suivi par une élimination.
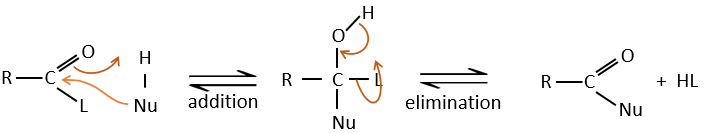
C’est ainsi la substitution d’un groupe par le nucléophile à la condition que le groupe L qui était auparavant sur le carbonyle soit un groupe partant meilleur que le nucléophile.
Cl–>RCOO–>RO–>OH–>NR2–>C–
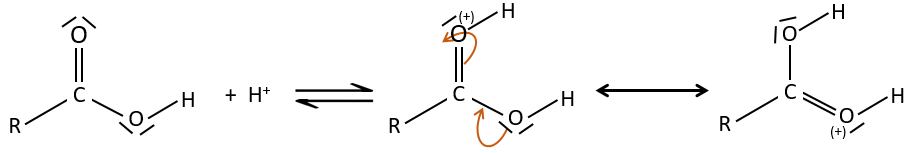
Comme pour les aldéhydes et les cétones la réaction peut être catalysée par un acide ou une base.
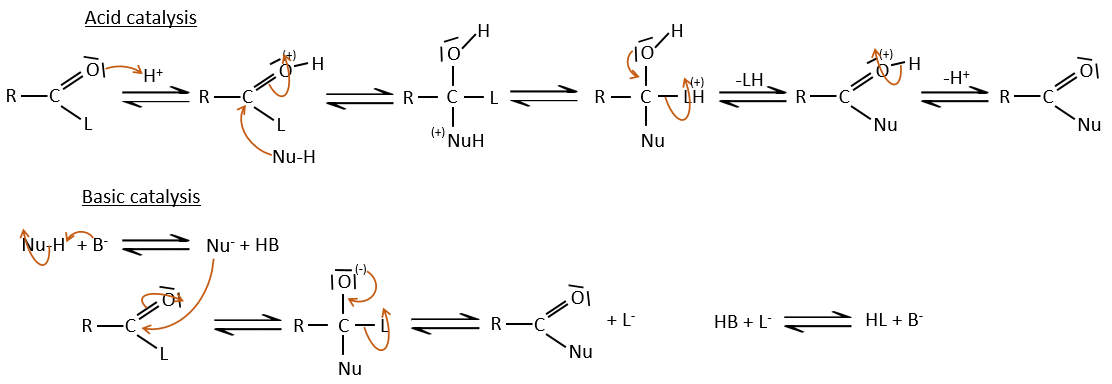
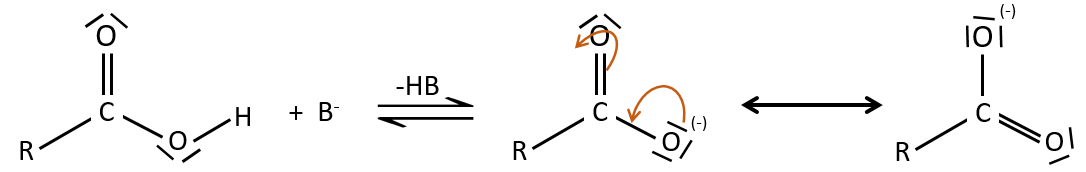
A noter que la réaction d’addition-élimination pour les acides carboxyliques est en concurrence avec les réactions acide-base. Le OH– n’est pas un très bon groupe partant et si le nucléophile est une base forte il y aura la formation de carboxylate. Dans le cas d’un nucléophile qui ne soit pas une base forte il y aura une concurrence entre les réactions AE et AB.
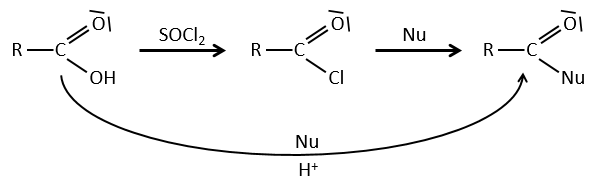
La formation du chlorure d’acyle :
D’après ce que nous avons écrit juste au-dessus nous ne pouvons pas remplacer RCOOH par RCOCl parce Cl– qui est un groupe partant meilleur que OH–. Cependant il y a un truc pour que cette réaction soit possible : nous utilisons du chlorure de thionyle (SOCl2). L’oxygène lié à l’hydrogène dans un groupe COOH est un nucléophile et peut attaquer l’atome de soufre en expulsant un atome de chlore qui attire le proton sur son chemin. L’acide chlorhydrique nouvellement formé peut maintenant réagir avec le thioester, éliminant une molécule de SO2 et de HCl.
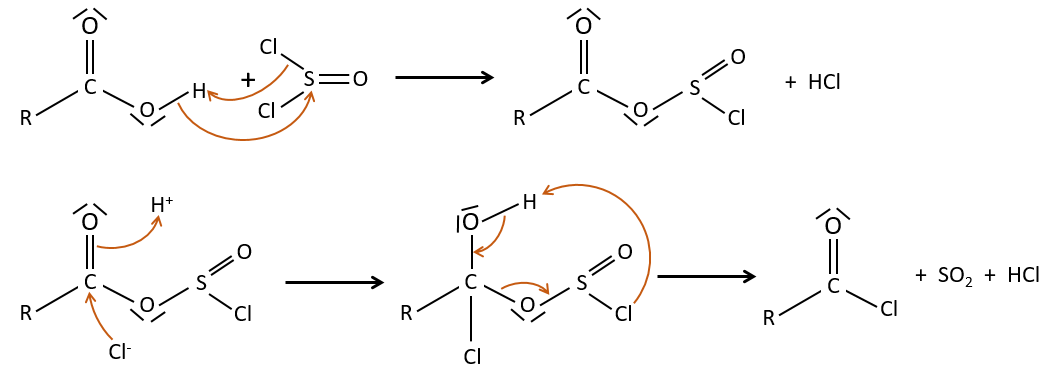
Le même genre de mécanisme est utilisé pour produire de l’acide bromique à partir des acides carboxyliques et PBr3 (de tribromure de phosphore). Pourtant ces réactions ne fonctionnent pas avec HCOOH parce que les produits HCOCl et HCOBr sont instables. Ils se décomposent en monoxyde de carbone et l’acide correspondant.
La formation d’esters :
Les acides carboxyliques réagissent avec des alcools pour produire des esters.
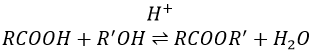
Les acides forts catalysent cette réaction.
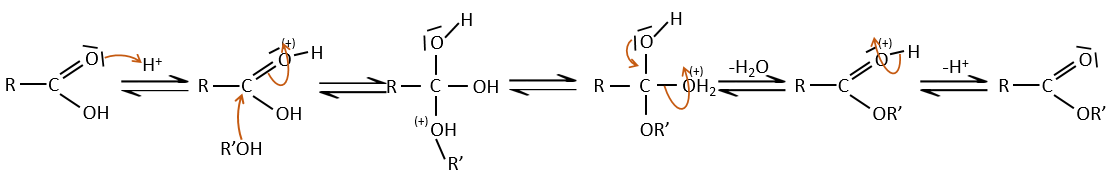
RO est un groupe partant légèrement meilleur que OH- si l’on veut déplacer l’équilibre vers la droite on le fait par l’utilisation d’un excès d’alcool ou d’acide carboxylique et par l’élimination de l’eau du système. Si nous voulons faire de l’hydrolyse d’un ester à savoir la réaction inverse, nous utilisons un excès d’eau. Il a été montré que l’hydrolyse des esters fonctionne de cette façon (attaque de l’eau sur le groupe carbonyle et pas d’attaque de l’eau sur R’) par l’utilisation d’isotopes de l’oxygène esterique.
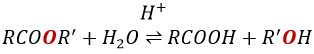
Une autre façon d’obtenir des esters est l’utilisation de diazométhane, la réaction que nous avons vu au début du chapitre de l’élimination.

Des estérifications intramoléculaires sont plus favorables que les intermoléculaires pour des raisons entropiques et sont particulièrement favorables si un cycle de 5 ou 6 atomes se forme.
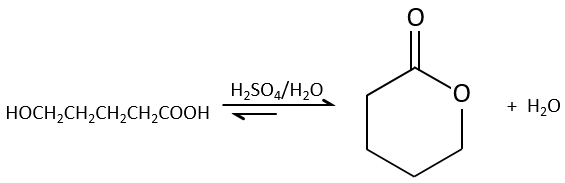
Ce type de cycle est appelé une lactone (δ-lactone si c’est un cycle de 6 atomes de carbone, γ-lactone si c’est un cycle de 5, etc.). Les lactones sont sensibles à l’hydrolyse.
Formation d’amides :
Dans des conditions normales une amine réagit avec un acide carboxylique pour former un sel (carboxylate d’ammonium) par une réaction acido-basique. Une fois que l’amine a pris le proton elle ne peut plus attaquer le groupe carbonyle.
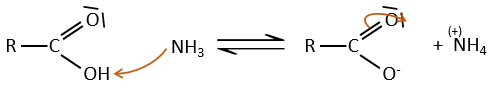
Si l’on chauffe le système, la réaction va plus en arrière et un autre processus, thermodynamiquement plus favorable se produit, conduisant à la formation d’un amide. Dans un amide l’atome d’azote est non basique.
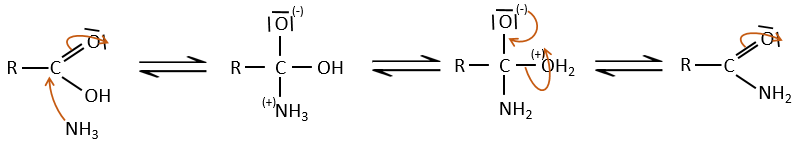
Les réactions intramoléculaires sont possibles et conduisent à la formation de lactames.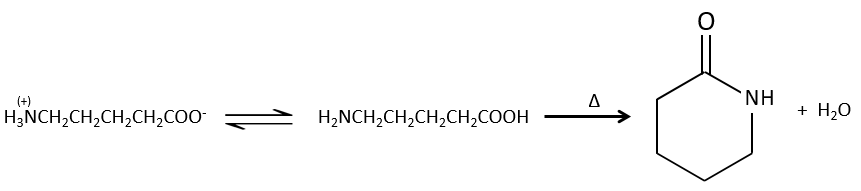
Les réactions d’acides carboxyliques et leurs dérivés :
Nous avons montré plusieurs réactions des acides carboxyliques. Il est souvent intéressant d’envisager de transformer l’acide carboxylique en un chlorure d’acyle avant la réaction souhaitée. Les chlorures d’acyle sont habituellement plus réactifs et cette étape supplémentaire dans le procédé peut augmenter le rendement global de la réaction.
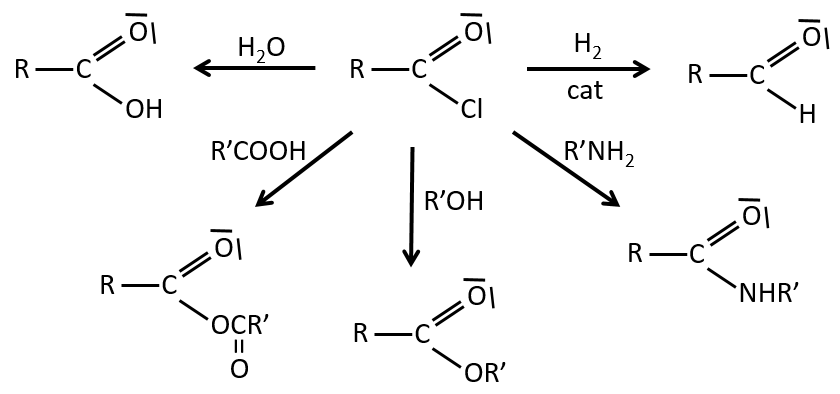
Les réactions des chlorures d’acyle :
Du chlorure d’acyle on peut obtenir beaucoup d’autres groupes fonctionnels.
Les réactions d’esters :
Comme les esters sont moins réactifs que les chlorures d’acyle, moins de réactions sont disponibles. On peut hydrolyser l’ester, échanger la chaîne estérique avec une autre chaine estérique ou générer un amide.
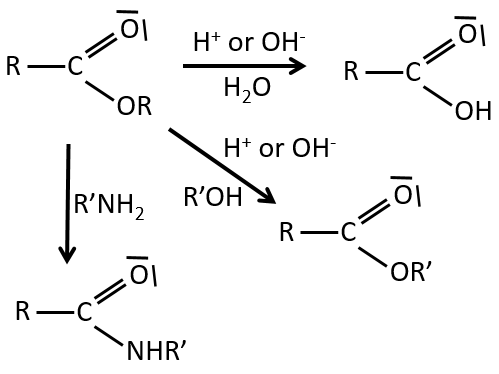
Une réaction supplémentaire consiste à utiliser un réactif de Grignard sur un ester pour obtenir un alcool tertiaire. La cétone est plus réactive que l’ester de sorte que la réaction se poursuit.

Les réactions des amides :
Les liaisons peptidiques peuvent être hydrolysées.
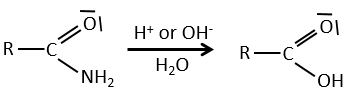
Réactions des nitriles :
L’atome de carbone à partir d’un nitrile est dans le même état d’oxydation que celui de l’acide carboxylique. Un nitrile en présence d’eau et d’acide conduit à la formation d’un acide carboxylique et le rejet de l’ammoniac.
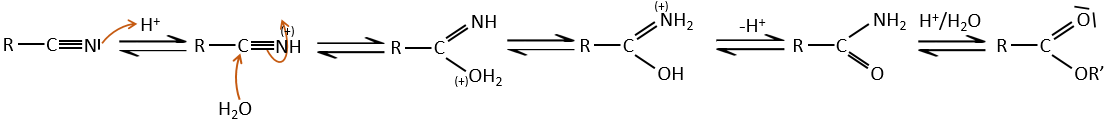
Les nitriles peuvent également réagir avec des réactifs de Grignard mais la réaction s’arrête à la formation de cétones.
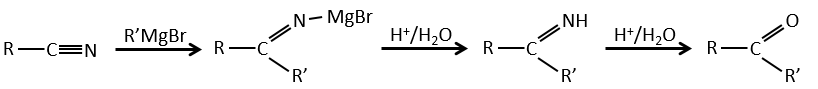
Les nitriles peuvent également être réduits en amines primaires par LiAlH4.
Chapitre 9 : carbanions en α du carbonyle
Dans la section précédente, nous avons discuté de la possibilité de réaction sur le groupe carbonyle des molécules organiques. Le carbone est un électrophile et l’oxygène est un nucléophile. Cependant la présence d’un groupe carbonyle peut impliquer d’autres processus. Les hydrogènes qui se trouvent sur un carbone voisin d’un groupe carbonyle, à savoir le carbone dans la position α du carbonyle, sont des acides et les bases fortes peuvent les arracher.
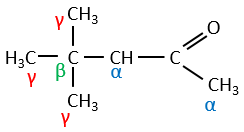
Son pKa est d’environ 20 (à titre de comparaison, l’atome d’hydrogène d’un groupe -C-H a un pKa de 40, un a=C-H a un pKa de 37 et -O-H a un pKa de 18). Le résultat est l’ion énolate. Il est stabilisé par résonance avec le carbonyle.
Its pKa is around 20 (for comparison, the hydrogen of a –C-H has a pKa of 40, a =C-H a pKa of 37 and –O-H a pKa of 18). The result is the enolate ion. It is stabilised by resonance with the carbonyl.
Tautomérisation :
L’ion énolate peut réagir par son atome d’oxygène ou l’atome de carbone en α.
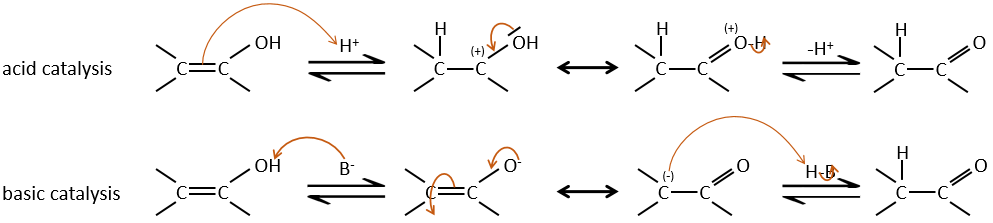
Les Énols sont généralement instables et tautomérisent dans le carbonyle correspondant. C’est la tautomérie énol-cétone. La tautomérisation se fait par catalyse acide ou basique.
La différence d’énergie est d’environ AG = 15kcal en faveur ou de la forme de cétone. L’équilibre énol-cétone peut être déplacé si une forme est stabilisée par la structure de la molécule.
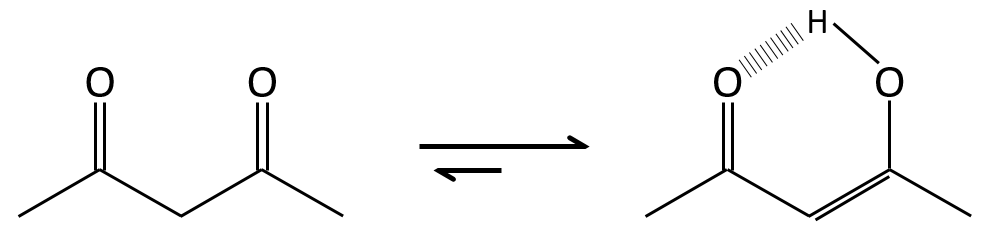
La tautomérisation permet de modifier la conformation de la molécule. Une molécule cis tautomérise pour devenir trans et diminue l’encombrement stérique.
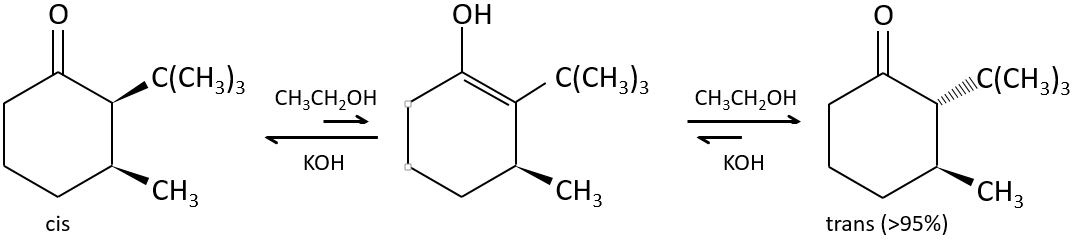
Nous pouvons également échanger l’hydrogène par ses isotopes en raison de la tautomérie.
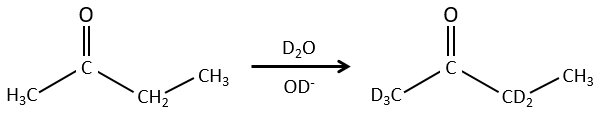
Les propriétés chimiques des isotopes sont presque identiques (parce que ces propriétés sont données par les électrons) mais certaines propriétés physiques peuvent être différentes entre les isotopes d’un même élément. La vitesse de réaction et la température d’ébullition sont deux exemples de propriétés qui changent en fonction de l’isotope. Il est ainsi possible de séparer les espèces portant des isotopes par distillation ou par centrifugation.
Halogénation :
L’hydrogène peut également être remplacé par des halogènes ou par le biais de catalyse de base acide. Dans ce cas, les résultats sont différents selon le catalyseur que nous utilisons.
– Catalyse acide: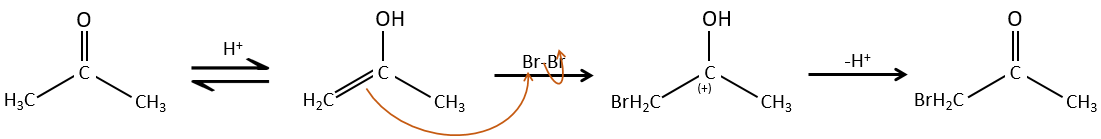
Une fois qu’un atome d’halogène est sur la molécule, il est plus difficile de former l’énol car il est plus difficile pour C = O d’attraper un proton en raison de l’effet inducteur-capteur de l’halogène.
– La catalyse basique:
Once one halogen is on the molecule, it is harder to form the enol because it is more difficult for C=O to catch a proton due to the inductive captor effect caoteue , of the halogen.
– La catalyse Basique:
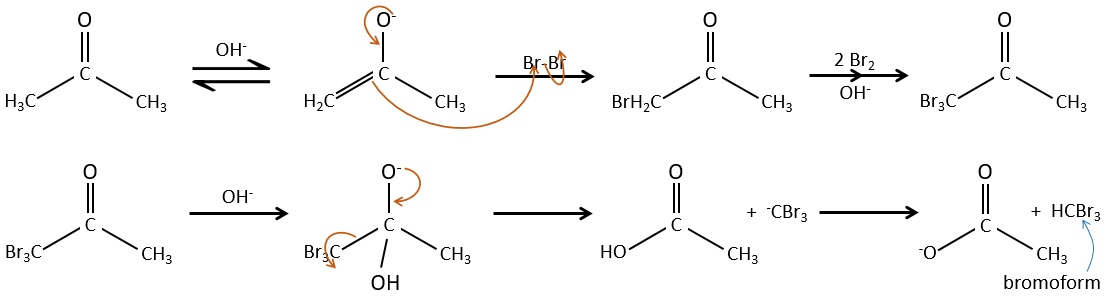
Au cours de la catalyse basique nous n’avons pas besoin de protoner le carbonyle et l’halogénation peut avoir lieu autant de fois qu’il y a des protons alpha.
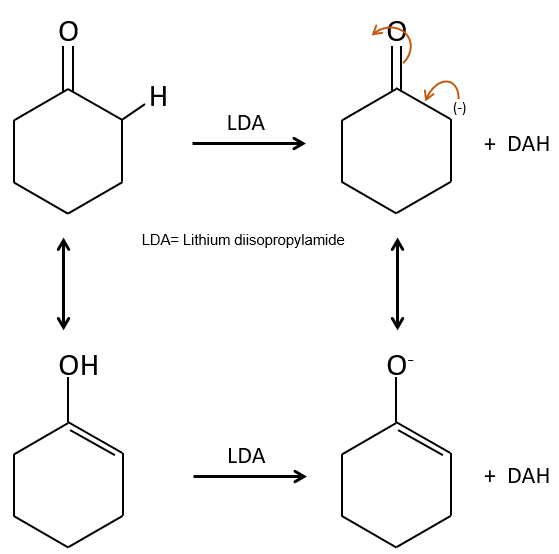
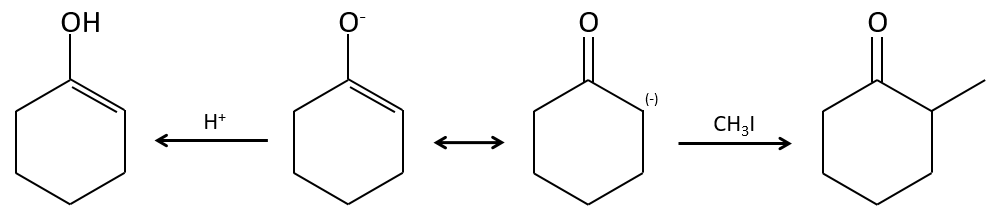
Alkylation :
Nous avons vu dans la section précédente que NAH seule ne réduit pas les carbonyles. Cependant il a un effet sur le carbone en α du carbonyle. C’est un bon moyen d’introduire un groupe alkyle à ce poste.
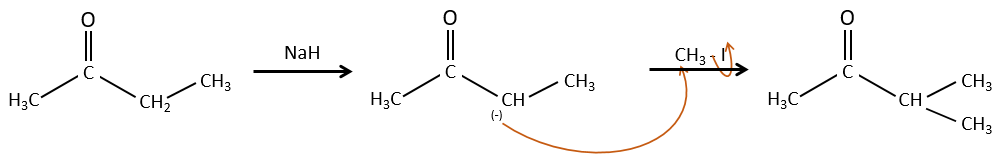
Une base forte peut aussi prendre le proto α de carbonyle.
Le problème c’est que la réaction peut se poursuivre s’il y a plusieurs hydrogènes en α, du carbonyle et polyalkylation est fréquente, ce qui conduit à un mélange racémique des produits.
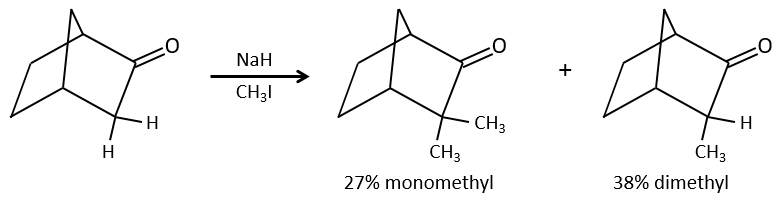
En outre la régiosélectivité est faible, ce qui donne une possibilité supplémentaire de produit.
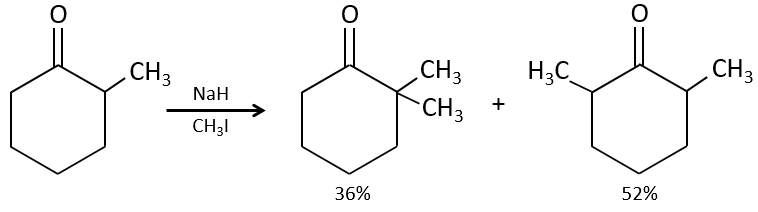
Pour éviter une polyalkylation nous pouvons remplacer l’oxygène par pyrrolidine (C4H9N) pour obtenir l’énamine correspondante qui a l’avantage d’un faible polyalkylation.
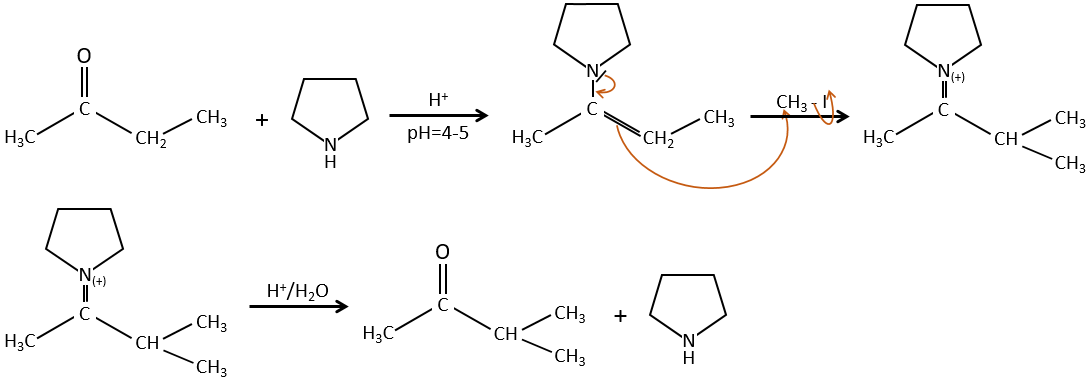
Cette protection est également efficace pour les aldéhydes qui sont soumis à la réaction d’aldolisation dans des environnements basiques.
La condensation aldolique :
Si nous ajoutons un peu de NaOH dans une solution d’acétaldéhyde des dimères sont formés. La première étape consiste en la déprotonation de l’ α de carbonyl par la base afin d’obtenir l’ion énolate qui attaque le prochain aldéhyde, conduisant à la formation d’un β-hydroxyaldéhyde. La β-hydroxyaldéhyde est stabilisée par sa liaison hydrogène interne.
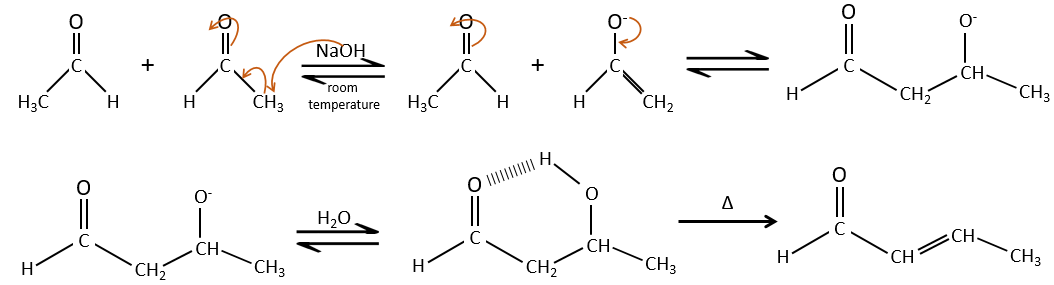
Si nous chauffons la solution, la molécule perd une molécule d’eau pour former une énone (la double liaison conjuguée avec un groupe carbonyle).
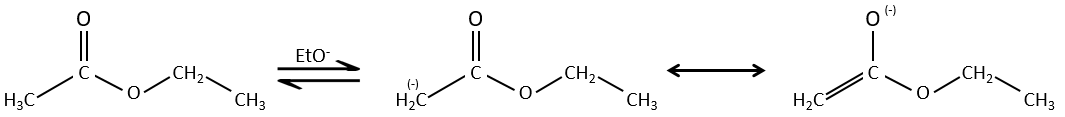
La condensation de Claisen :
La réaction analogue pour les esters est appelé la condensation de Claisen. NaOH n’est pas utilisé dans le cas présent parce que cela donnerait un clivage de l’ether. Nous pouvons utiliser par exemple l’éthylate de sodium pour obtenir l’énolate.
Comme dans la condensation aldolique l’ion énolate attaque un autre ester pour obtenir des ß-cétoesters.
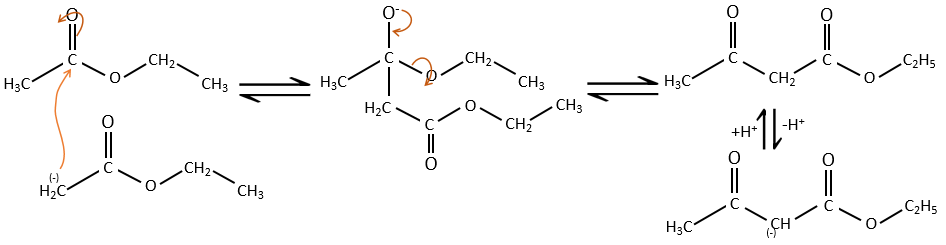
La condensation de Claisen peut être réalisée avec deux esters différents (la condensation mixte de Claisen) ou entre deux esters dans la même molécule (la condensation intramoléculaire de Claisen).
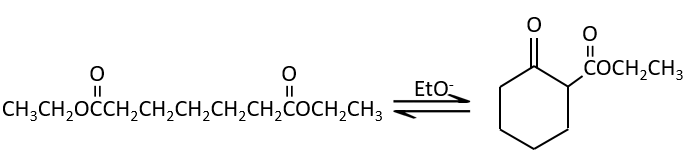
La réaction intramoléculaire est favorisée par rapport à une réaction intermoléculaire car elle ne diminue pas l’entropie.
Les propriétés des aldéhydes insaturées α,β et des cétones :
Ces espèces sont stabilisées par résonance.
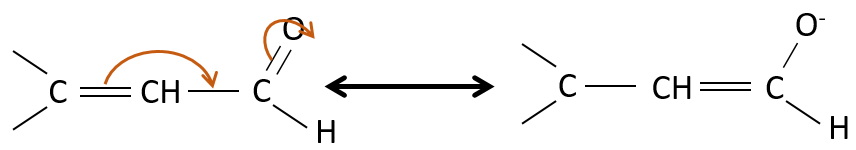
Ies cétones sont beaucoup plus stables que les aldéhydes, les cétones ß,γ-insaturés qui se réarrangent en composé α, β insaturé en présence d’une base.
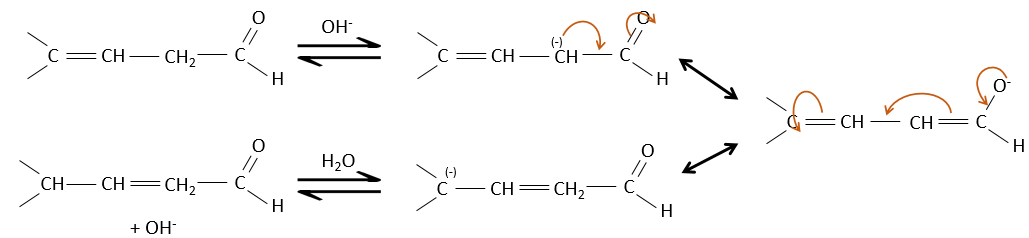
De toute évidence, la double liaison et les groupes carbonyles peuvent passer par des réactions de façon indépendante, tels que des réactions d’addition par exemple.
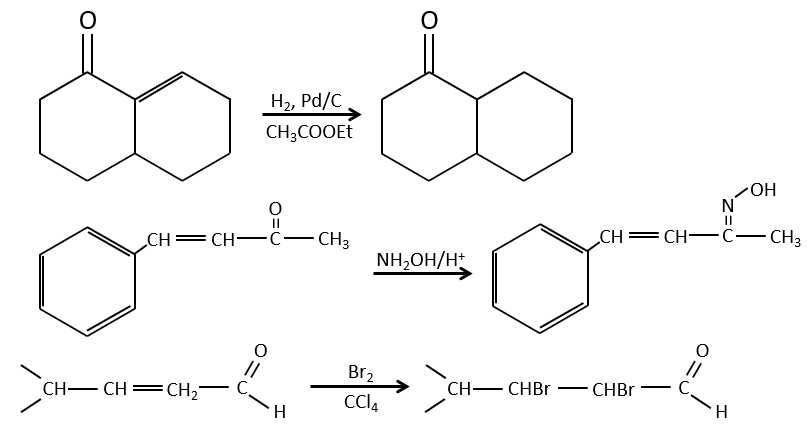
Mais les additions peuvent également être réalisées dans plusieurs cas sur les systèmes conjugués. Ce sont les additions 1,4.
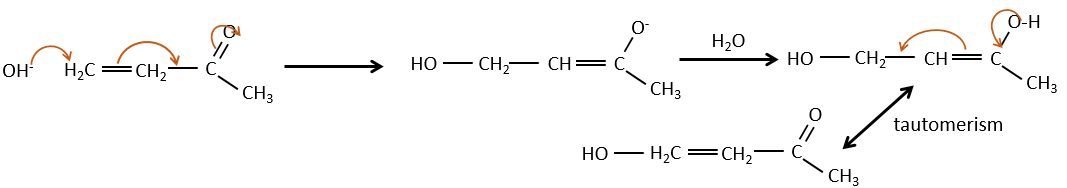
L’addition de Michael peut être suivie d’une condensation aldolique intramoléculaire, la réaction est connue sous le nom d’annulation de Robinson.
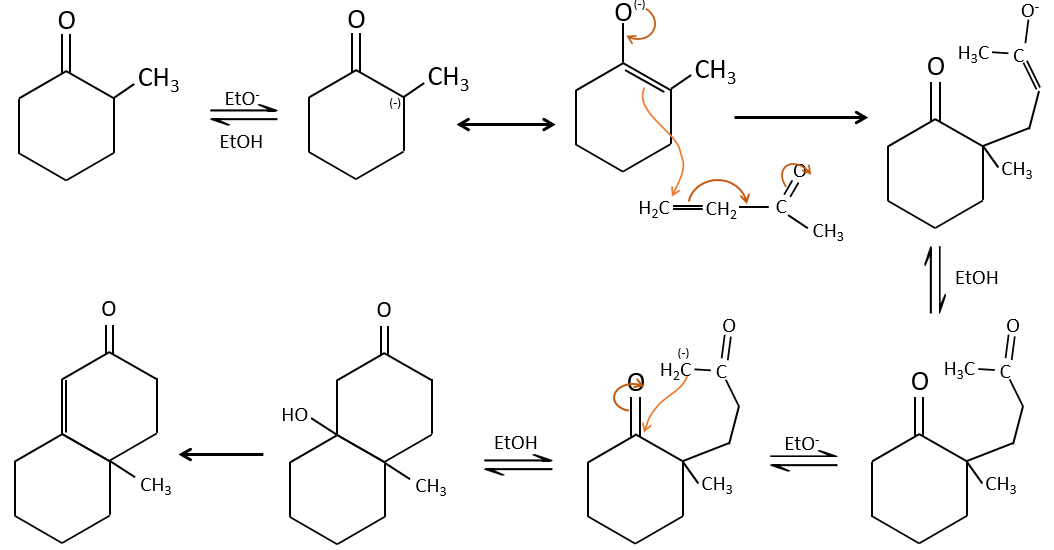
Chapitre 8: Réactions de l’élimination
Une réaction d’élimination est une réaction au cours de laquelle une molécule perd deux fragments A et B en tant que substrat neutre AB.
Les deux fragments A et B qui sont enlevés peuvent être retirés du même carbone, dans ce cas nous parlons d’une élimination 1,1, de deux atomes de carbone adjacents (élimination 1,2 ou élimination bêta) ou deux atomes de carbone séparés par un atome de carbone (élimination 1,3). L’élimination 1,2 est la plus commune.
L’élimination 1,1 :
Les deux fragments sont retirés d’un seul carbone. Elle conduit à la formation d’un carbène très réactif.
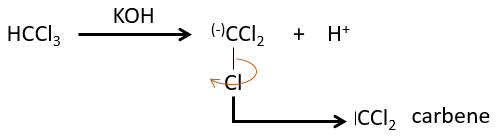
Cette réaction est possible parce que les atomes de chlore prennent les électrons de l’atome de carbone. A cause de cela, la liaison CH est déstabilisée et une base forte peut enlever le proton.
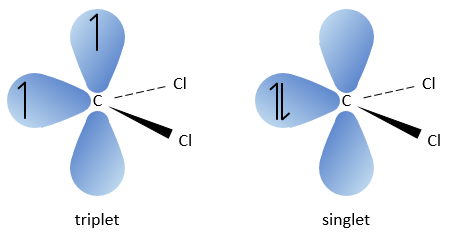
Le carbone du carbène est un singulet et est moins stable que un triplet. Il sera donc réagir rapidement, avec des liaisons doubles, pour obtenir une chaîne de cyclopropane. Un triplet ne serait pas en mesure d’attaquer la liaison π.
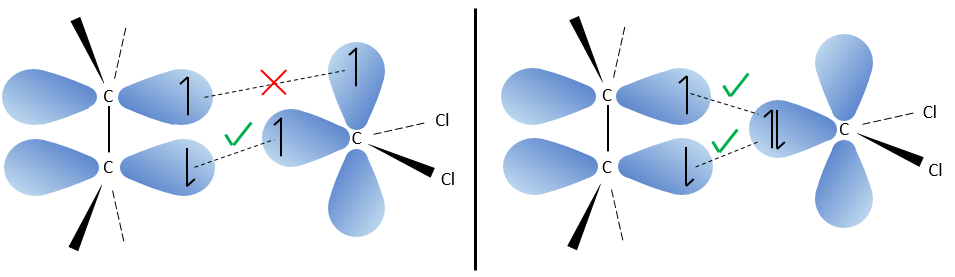
La réaction est stéréosélective (sur le même côté du plan).
Le plus petit est le carbène méthylène qui peut être produit à partir de diazométhane (vérifier nom anglais). Le diazométhane est explosif et toxique de sorte que la réaction doit être contrôlée.
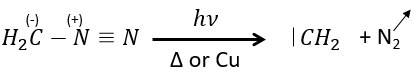
Il peut également être utilisé pour produire des esters méthyliques.
L ‘élimination 1,2 :
Ce type d’élimination conduit à la formation de nouvelles liaisons π. Il est à l’opposé d’une réaction d’addition. Nous discuterons seulement de la réaction principale c-à-d la β élimination mais il peut y avoir d’autres mécanismes impliqués pour obtenir une telle réaction.
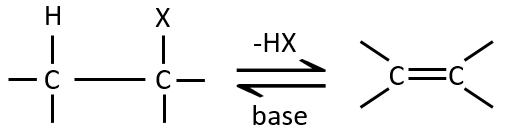
Comme pour les substitutions, où il ya un mécanisme avec une cinétique d’ordre 1 (SN1) et une d’ordre 2 (SN2), il y a une élimination d’ordre 1 (E1) et d’ordre 2 (E2).
Le mécanisme E1 :
La première étape de la réaction est le départ d’un anion et la formation d’un carbocation. Cette étape est lente et détermine la vitesse de la réaction.
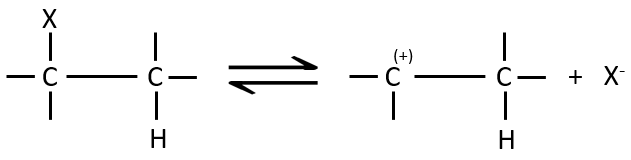
La deuxième étape est la capture d’un proton sur l’atome de carbone adjacent par une base. Il conduit à la formation de la liaison π pour avoir une espèce neutre.
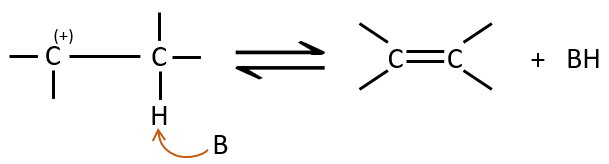
Nous pouvons savoir si une élimination est de l’ordre 1 à partir des éléments suivants :
1- l’ordre de la cinétique de la réaction (ordre 1).
2- l’effet isotopique : si l’hydrogène est remplacé par un deutérium, nous ne voyons pas de modification de la vitesse de réaction. Il en est ainsi parce que l’hydrogène ne participe pas à l’étape de détermination. Si elle a été impliquée, la vitesse serait divisé par 5 à 8.
![]()
3- la stabilité du carbocation : la stabilité du carbocation (degré de substitution ou hyperconjugaison) influe sur la vitesse de réaction.
4- les réarrangements: expliqués par l’existence d’un carbocation.
Mécanisme E2 :
La cinétique est d’ordre 2 et implique une réaction en une seule étape, où la prise de proton par la base et le départ du groupe partant sont simultanés.
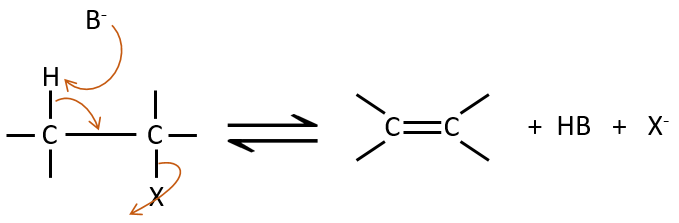
Cette réaction est possible seulement si le proton et le groupe partant sont en positions anti.
Les arguments en faveur du mécanisme E2 sont :
– Une cinétique d’ordre 2
– Un effet isotopique énorme
– Pas de réarrangement
– Énorme influence de l’énergie de la liaison C-X (X est le groupe partant)
La compétition entre les mécanismes :
En ce qui concerne les substitutions nucléophiles il existe une compétition entre les deux mécanismes. Ce n’est pas toujours 100% E1 mais plutôt un mélange des deux pour aboutir finalement un mélange racémique des produits. La force de la base et l’encombrement stérique ifluencent le mécanisme d’élimination utilisé : une base forte favorise le mécanisme E2 alors qu’une base faible et un encombrement stérique favorisent le mécanisme E1.
Or, les molécules présentant un bon groupe partant peuvent souvent être aussi l’objet d’une réaction de substitution en présence d’agents nucléophiles. Il existe donc une compétition entre E1, E2, SN1, SN2 et éventuellement d’autres réactions. Les trois principaux paramètres, à prendre en considération pour déterminer laquelle des réactions doit se produire, sont la force de la base, sa nucléophilie et l’encombrement stérique.
Les nucléophiles qui sont mauvais nucléophiles ont de bons rendements avec le SN2 avec des substrats primaires et secondaires. Si l’encombrement stérique est plus grand, le produit principal devient SN1. Si le nucléophile est une base forte, le produit principal est SN2 mais si l’encombrement stérique augmente, l’élimination devient plus importante.
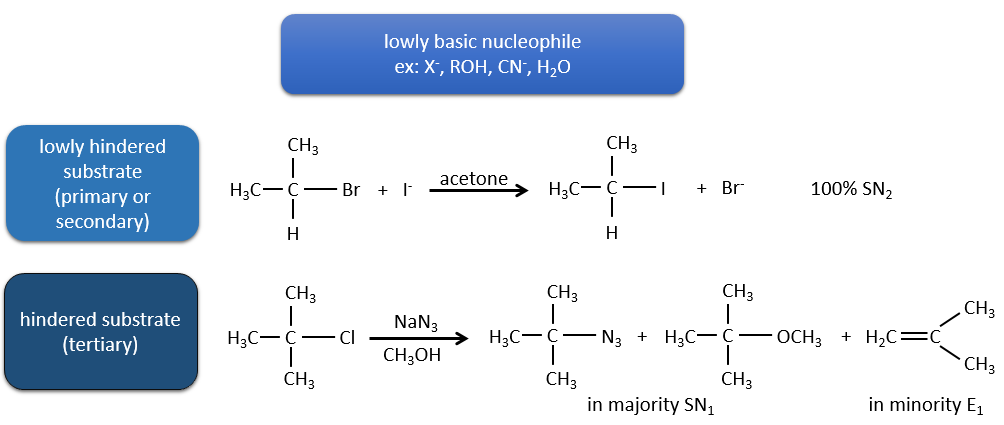
Les nucléophiles qui sont des bases fortes et qui ont un encombrement stérique favorisent l’élimination.
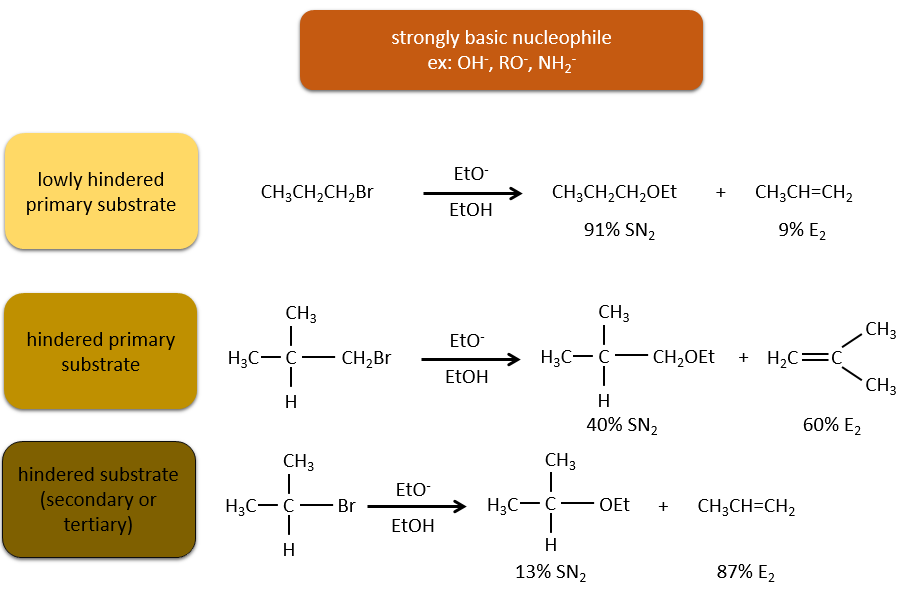
En resumé :
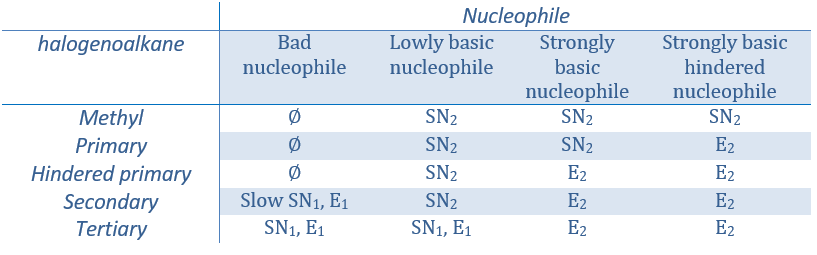
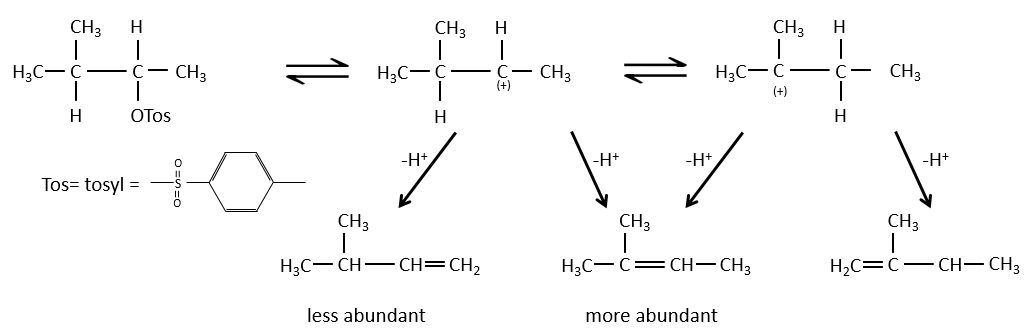
Régiochimie de l’élimination :
Lorsque le carbone portant le groupe partant n’est pas à une extrémité d’une chaîne, il peut y avoir plusieurs produits possibles. Nous pouvons prédire lequel des hydrogènes sera supprimé.
Si la double liaison générée peut être conjuguée avec d’autres liaisons π, ces produits seront favorisés.
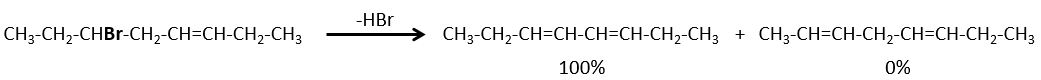
Dans le cas de substrats avec ponts (pontés), le produit n’engage jamais une liaison π sur une tête de pont.
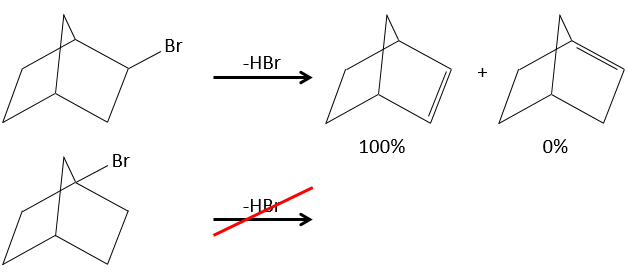
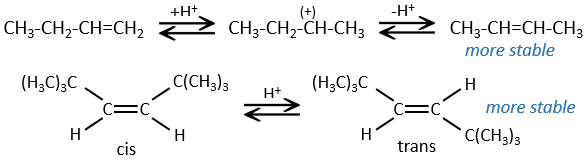
Pour une élimination E1 : l’élimination est déterminée par la stabilité des oléfines formées. Le plus substitué C = C est ainsi favorisé. C’est la règle de Zaitsev.
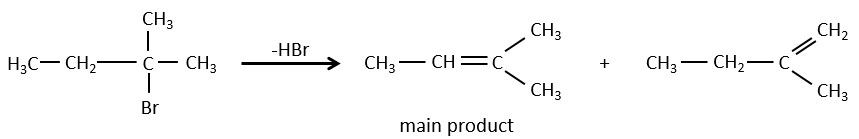
Il peut y avoir des exceptions en raison de l’encombrement stérique
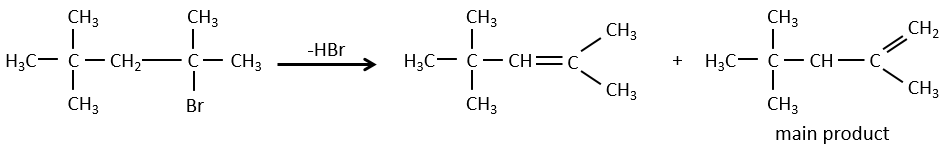
pour les éliminations E2, une H en anti est requis
la règle de Zaitsev est suivie si le groupe partant n’est pas chargé (la plus substituée C = C)
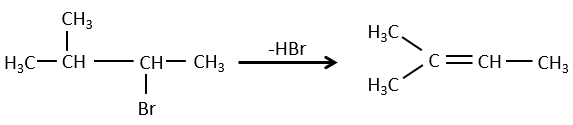
C’est le contraire si le groupe partant est chargé : c’est le règle de Hofmann
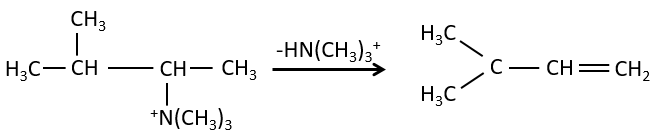
dans une telle élimination, c’est le proton le plus acide qui est pris par la base, à savoir l’atome de carbone le plus substitué car il est stabilisé par l’effet donneur mésomérique de la chaîne alkyle .
il peut être un bon moyen pour déterminer la position d’un atome d’azote dans une molécule pour voir si elle est primaire, secondaire, etc.
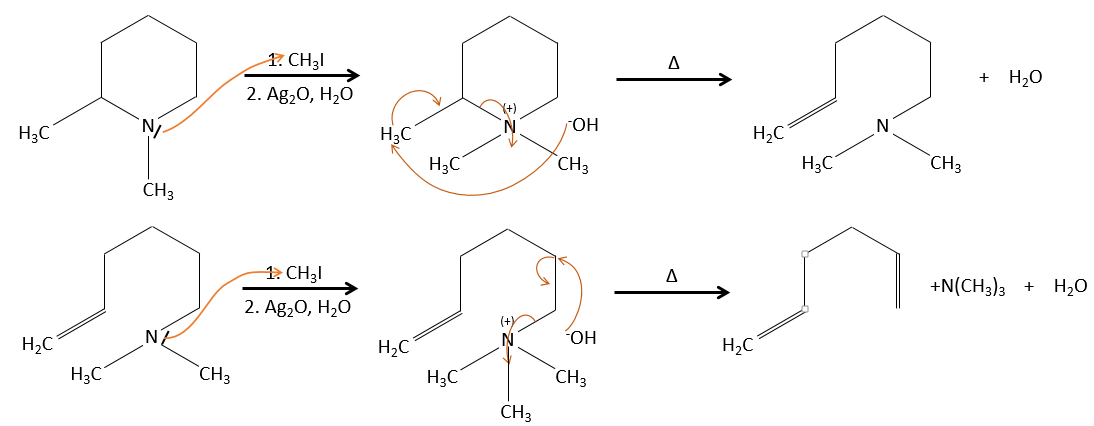
La configuration du produit :
Dans le cas d’une élimination E2, la réaction est stéréospécifique et il y a donc seulement une seule configuration possible.
Dans le cas d’une élimination E1, le produit est plus stable si les substituants volumineux sont en trans.
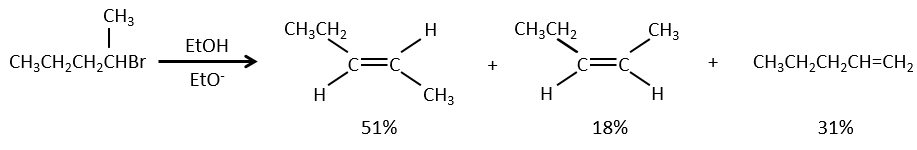
La déshydratation des alcools :
Cette élimination ne se produit pas dans des environnements basiques ou neutres : OH– est un mauvais groupe partant et nous aurons former –O– en présence d’une base forte. Dans un environnement acide, l’élimination peut se faire par E1 ou par E2 . Le E1 est favorisé dans le cas de produits substitués et E2 est le principal mécanisme de carbones primaires.
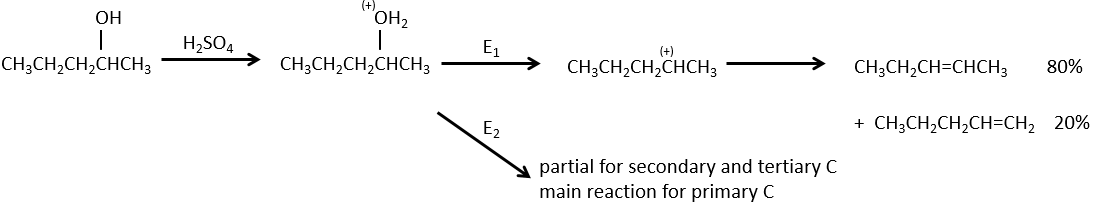
Réactivité : tertiaire> secondaire> primaire
E1 E1 E2
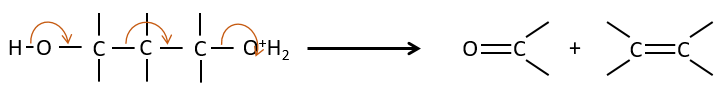
La fragmentation de 1,3 diols :
C’est un cas d’ élimination 1,3 au cours de laquelle la molécule est clivée
Chapitre 7 : Additions électrophiles sur les alcènes et alcynes
L’intérêt des ajouts (additions) électrophiles sur les liaisons C = C est de transformer deux carbones sp2 en deux carbones sp3 et d’ajouter une chaîne ou un groupe sur la molécule existante.
Comme deux atomes de carbone sont impliqués dans la réaction, l’ajout du nouveau groupe peut donner plusieurs produits avec plus ou moins de (stéréo) sélectivité. En outre la transformation en carbones sp3 introduit également une régiosélectivité qui doit souvent être prise en considération.
Par conséquent nous allons discuter dans cette section les méthodes et les règles qui doivent être suivies pour obtenir le produit souhaité au cours de l’addition d’un groupe électrophile sur les liaisons doubles ou triples.
L’hydrogénation catalytique :
Il est, en principe, l’ajout (l’addition) le plus facile sur une liaison C = C car il n’y a pas de nouveau groupe sur le produit. Les carbones sp2 sont réduits en carbones sp3. Or cette réaction n’est pas spontanée en dépit du fait que c’est une réaction exothermique. L’énergie d’activation pour briser la liaison H-H est énorme et un catalyseur est nécessaire pour effectuer la réaction.
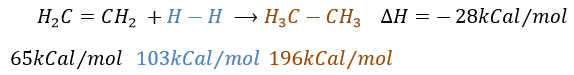
Comme catalyseur on peut utiliser le platine (Pt), le nickel de Raney(Ni) et du palladium sur C (Pd/C).
Ils ont la capacité de dissocier les atomes d’hydrogène et de les fixer sur leur surface ce qui les rend disponibles pour l’ajout sur les alcènes. Comme nous avons vu dans la section de la cinétique, le catalyseur diminue l’énergie d’activation de la réaction et le processus peut être représenté en 4 étapes :
– Approche et la liaison des réactifs
– Déplacement sur la surface
– Réaction(s)
– Départ des produits
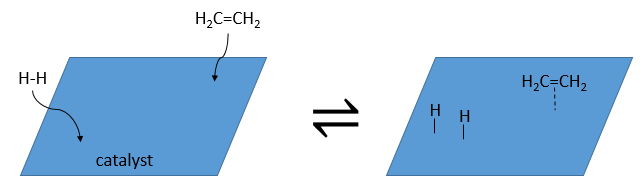
Lors de la première étape les réactifs ont lieu sur la surface du catalyseur hétérogène. La quantité de réactif qui peut avoir lieu sur le catalyseur dépend de sa surface. Il est donc conseillé d’utiliser de petites particules de catalyseur pour augmenter le rapport surface/volume (ou surface/masse). Les alcènes se placent parallèlement à la surface dans la direction de la liaison π.
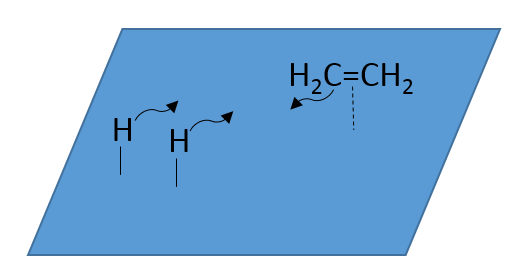
Il n’y a aucune raison que les réactifs se lient immédiatement sur des spots à proximité. La deuxième étape consiste donc à ce que les réactifs se déplacent sur la surface. Ils finiront par être en contact les uns avec les autres de sorte que la réaction peut avoir lieu.
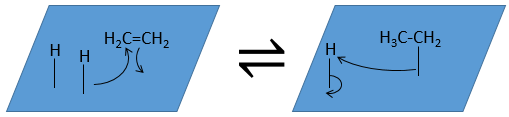
La troisième étape (et éventuellement les suivantes) est la réaction entre les réactifs. Un hydrogène activé attaque un carbone sp2 tandis que la liaison π relie le second carbone avec la platine. La prochaine étape de la réaction est que la liaison entre le carbone et le platine attaque l’hydrogène restant.
Durant la dernière étape, l’alcane quitte la surface du catalyseur en laissant un espace pour que les réactifs se lient.
L’hydrogénation est stéréosélective, à savoir qu’au niveau de la liaison C = C la place des groupes sur les atomes de carbone ne sont pas équivalents et la molécule est plane. Si les atomes d’hydrogène sont fixés sur le même côté (mais pas sur le même atome de carbone) de la molécule alors c’est une addition syn. Comme les groupes ajoutés sont des hydrogènes , le produit est donc cis (voir substitutions nucléophiles et cis et trans des produits).
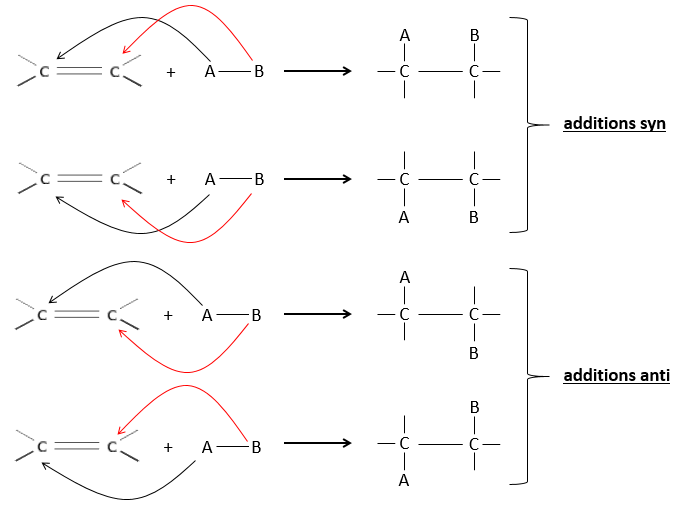
Si les groupes ont été ajoutés sur les côtés opposés des molécules (toujours pas sur le même carbone) l’addition serait anti et le produit serait trans. Ce produit ne sera pas observé parce que les réactifs sont liés au catalyseur et ne peuvent pas se déplacer librement pendant la réaction. Les deux hydrogènes sont ainsi du même côté de la molécule avant la réaction et après la réaction.
Ceci est important seulement si, après la réaction, le carbone sp3 nouvellement formé ne peut pas tourner. Ce sera le cas lorsque la liaison est impliquée dans un cycle ou s’ il y a des groupes volumineux. En outre le fait que l’on obtient seulement le produit cis ne signifie pas qu’un seul produit est possible. La raison en est qu’il y a un seul plan de chaque côté de la liaison de π (pour un total de 2). Si les deux plans sont équivalents, nous obtenons un mélange racémique 50-50 de deux produits cis.
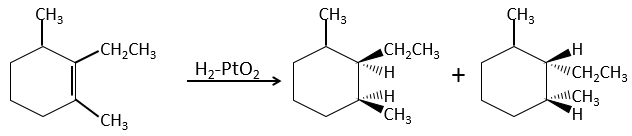
Si les plans ne sont pas équivalents un des produits est favorisé : les atomes d’hydrogène sont fixés sur le côté moins occupé parce que les alcènes se placent sur le catalyseur de sorte que les groupes volumineux soient loin du catalyseur.
Les catalyseurs solubles peuvent également être utilisés pour cette réaction. C’est le cas pour le catalyseur de Wilkinson (tris(triphénylphosphine) rhodium (I).
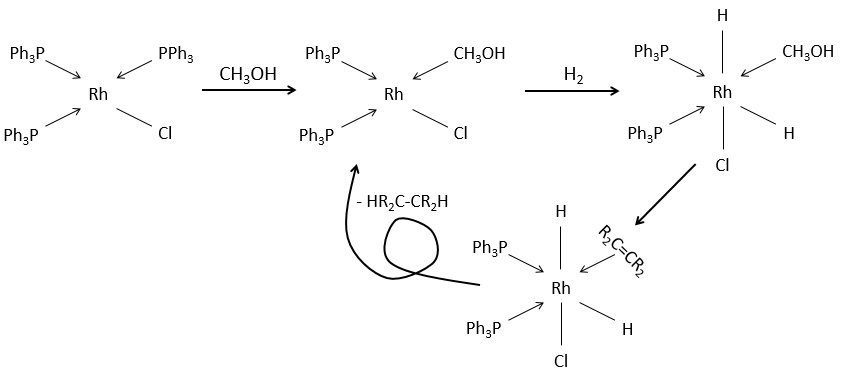
Il conduit également aux produits cis.
En termes de réactivité les atomes de carbone substitués réagissent plus lentement que les carbones ayant moins de groupes en raison de l’encombrement stérique : les atomes d’hydrogène ne sont pas très mobiles sur la surface du catalyseur et la liaison π doit être proche pour obtenir l’hydrogénation.
Les alcynes réagissent aussi plus rapidement que les alcènes ce qui permet d’hydrogéner tous les alcynes en alcènes avant la formation des alcanes. Nous pouvons ainsi limiter l’hydrogénation des alcènes si nous utilisons un mauvais catalyseur et si nous mettons un équivalent de H2 par alcyne. Nous allons obtenir l’alcane avec 2 équivalents de H2 par alcyne.
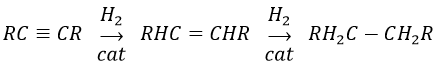
Encore une fois cela conduira à l’alcène cis. Pour obtenir l’alcène trans nous utilisons du sodium dans l’ammoniac. Le sodium génère un radical sur un atome de carbone et une charge négative sur l’autre.
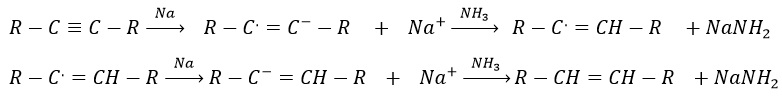
Le produit trans est favorisé pour des raisons stériques.
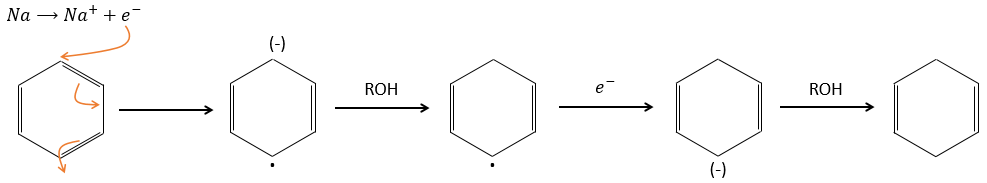
Les produits aromatiques peuvent difficilement être hydrogénés avec H2/cat (à 150° C), mais ils peuvent également être réduits différemment par la réduction de Birch impliquant Na, NH 3 et un alcool.
Autres types d’ajouts électrophiles :
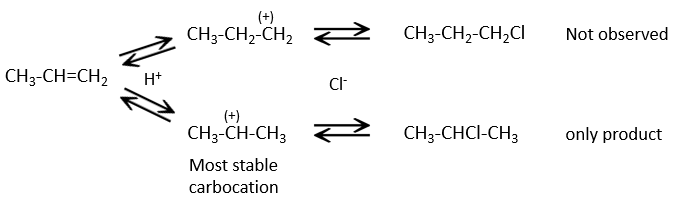
L’addition de HX :
Les autres réactifs HX contenant un atome d’hydrogène peuvent être plus facilement cassés (ou sont déjà ioniques) et ne nécessitent pas de catalyseur. Dans le cas d’un acide halogène, en raison de leur caractère nucléophile, les liaisons de π qui sont plus faibles que les liaisons σ ont tendance à attaquer les molécules électrophiles. Dans ce cas la cible électrophile est le proton. Il est appelé une attaque électrophile. Le X nucléophile attaque le carbone positif (ou carbocation) par la suite.
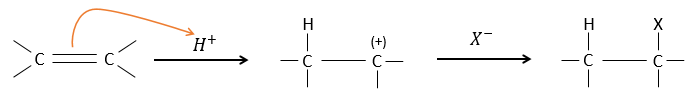
Cette réaction ne comporte pas de stéréosélectivité parce qu’il y a deux étapes distinctes mais implique la régiosélectivité : c’est la règle de Markovnikov qui dit que l’halogène est fixé sur le carbone le plus substitué ou, inversement que le proton, est fixé sur le carbone le moins substitué. Personnellement je préfère rappeler que le carbocation le plus stable est formé. Rappelez-vous que les carbocations peuvent réorganiser la molécule pour former le carbocation plus stable.
L’addition d’eau :
L’eau est ajoutée à alcènes en présence d’acide sulfurique :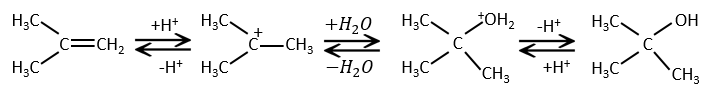
L’acide sulfurique est choisi parce que le sulfate est moins nucléophile que l’eau (parce que la charge est stabilisé par résonance). HCl doit être évité car Cl– est plus nucléophile que H2O. En outre toutes les étapes sont réversibles. Cela signifie que l’alcool nouvellement généré peut être déshydraté dans le cas d’un excès d’acide.
La présence d’un carbocation explique quelques réarrangements d’alcènes qui peuvent être observés dans des solvants acides.
L’addition de X2 :
Des halogènes sont additionnés à C = C pour obtenir des produits trans.
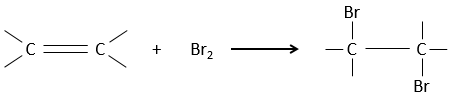
Il est étrange que des éléments aussi chargés en électrons que des halogènes soient utilisés comme électrophiles pour cette réaction. La raison en est que la liaison entre les halogènes est polarisable. Un halogène va donc ressentir les électrons de la liaison π et va transférer une partie de ses électrons à l’autre halogène. L’halogène partiellement positif peut donc être attaqué par la liaison π et forme un halonium intermédiaire qui sera attaqué par l’halogène négatif sur l’autre côté de la liaison C-C.
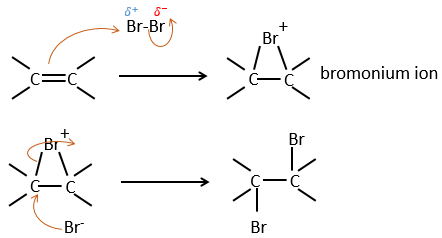
Cette réaction est stéréosélective (toujours trans) et stéréospécifique (le résultat dépend de la disposition du réactif). En présence d’autres agents nucléophiles, tels que l’eau, la réaction commence normalement mais il existe une compétition entre X– et le nucléophile.
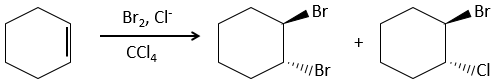
Cette réaction est plus lente sur alcynes parce que le halonium intermédiaire est moins stable. L’halogénation est donc difficile à contrôler : les alcènes réagissent plus vite que les alcynes.
La réaction d’oxymercuration :
Cette réaction est un moyen utile pour former des alcools ou des éthers à partir d’alcènes. Elle implique l’acétate de mercure et de l’eau/alcool dans le THF comme solvant. L’ouverture nucléophile de l’eau/alcool se fait dans la position anti et sur le carbone le plus substitué (il n’y a donc plus d’espace pour l’acétate de mercure).
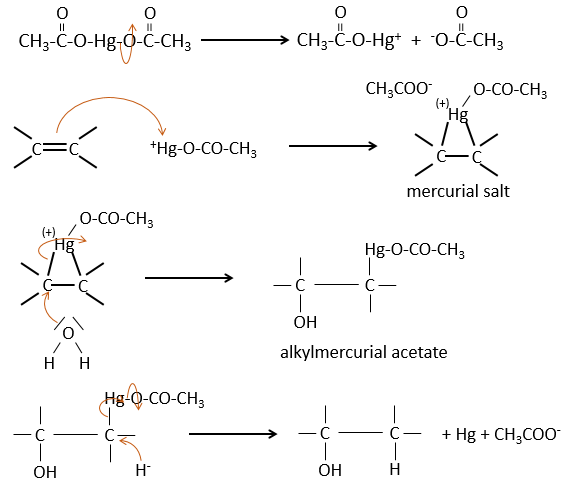
L’oxymercuration suit la règle de Markovnikov mais il n’y a pas de carbocation impliqué dans le processus. Comme tous les ajouts il est régiosélectif.
L’eau conduit à la formation d’un alcool et on peut utiliser un alcool au lieu de l’eau pour obtenir un éther.
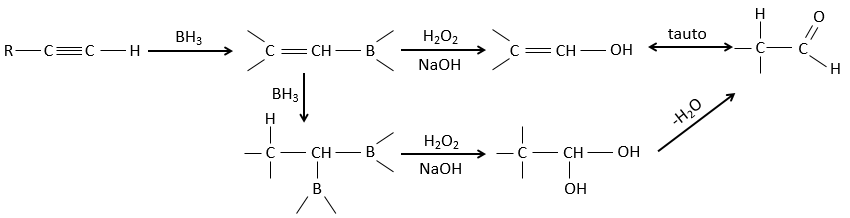
Réaction d’hydroboration :
BH3 (borane) est un acide de Lewis qui est stable sous la forme d’un dimère et peut remplacer son hydrogène en 3 par 3 chaînes de carbone de THF (tétrahydrofurane (CH2)4O). Une fois que les chaînes sont liées, il est possible de supprimer le bore par une oxydation avec H2O2, NaOH et H2O2 pour obtenir le plus grand nombre d’alcools.
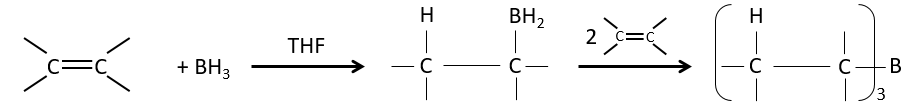
Cette réaction ne suit pas la règle de Markovnikov (anti-Markovnikov) et est stéréosélective. Le mécanisme de la réaction implique l’orbital p vide du bore qui interagit avec la liaison π pour former un complexe. Ce complexe permet un état de transition, avec 4 centres, dans lequel les liaisons se déplacent.
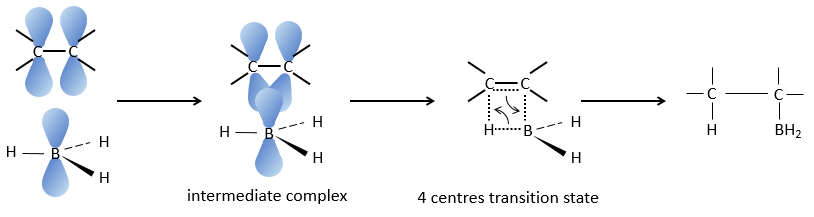
La règle Markovnikov s’applique lorsque l’hydrogène est chargé positivement. Dans le cas présent nous ajoutons H– et BH2+.
L’élimination du bore est fait avec rétention de configuration et est stéréospécifique. Le bore est moins électronégatif que le carbone et donne ainsi ses électrons. NaOH prend un proton du H202 pour former HOO– qui attaque le bore, transferant sa charge négative. Il existe un réarrangement qui met un atome d’oxygène entre la chaîne et le bore.
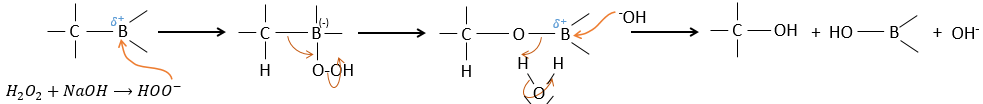
L’eau et OH– agissent à l’étape suivante pour supprimer le bore.
Cette addition peut également se faire sur des alcynes pour former des aldéhydes par tautomérisation.
La réaction des alcènes avec des acides peroxycarboxyliques
Les acides peroxycarboxyliques sont généralement instables. Le MCPBA et MMPP sont toutefois des peroxyacides stables et sont largement utilisés dans les laboratoires :
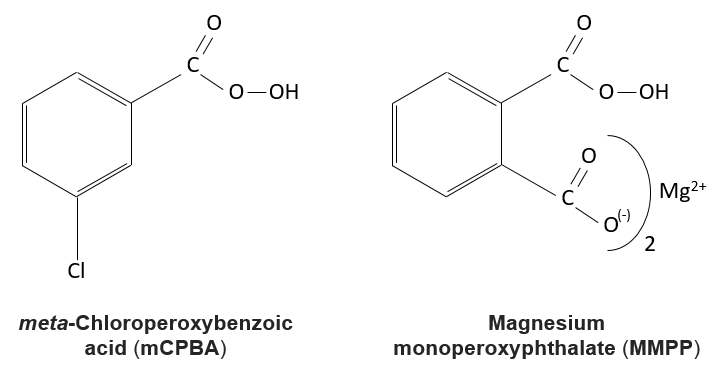
Un des oxygènes des acides peroxycarboxyliques est électrophile. Ces acides peuvent donc réagir avec des alcènes pour former des époxydes et de l’acide carboxylique.
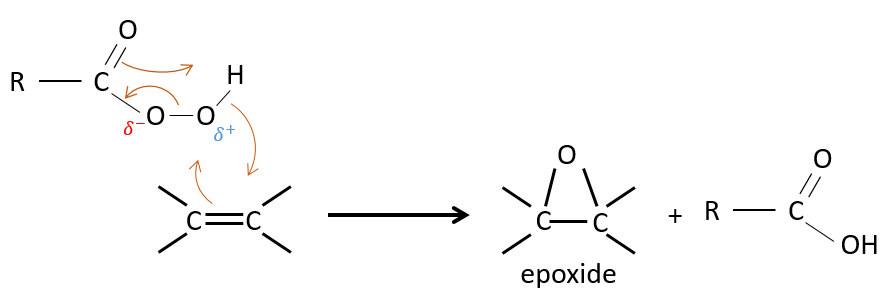
L’addition est donc une réaction concertée, est stéréospécifique et est syn. La réaction est faite dans le plan le moins encombré de l’alcène. La réactivité augmente avec la quantité de substituants qui partagent leurs électrons avec la liaison π (les chaînes alkyles par exemple).
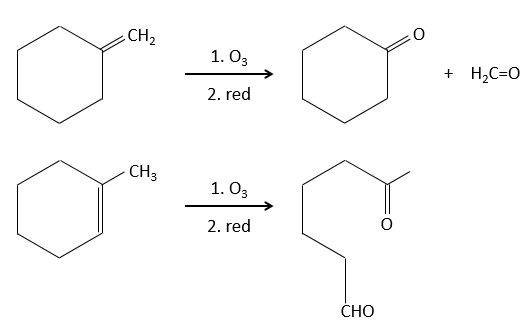
L’ozonolyse :
L’ozone peut briser des liaisons doubles. Dans un premier temps il ouvre la double liaison pour donner un intermédiaire appelé molozonide.

Il rompt pour former l’ozonide qui peut être la prochaine oxydé ou réduit.
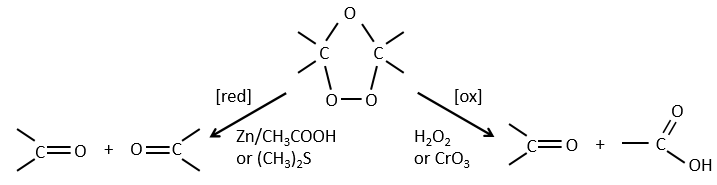
L’ozonolyse est donc un bon moyen pour obtenir les cétones et les acides carboxyliques à partir d’alcènes. La méthode est également utile pour déterminer la position d’une double liaison dans une molécule quand nous ne pouvons pas facilement la déterminer avec les techniques habituelles.
Chapitre 6: Les polymères
Aujourd’hui on trouve des polymères partout.Ils peuvent être solides ou flexibles, résistants à des températures basses ou élevées mais sont généralement plus légers que les matériaux qu’ils remplacent. Par exemple, des chaises en bois ou des chassis métalliques sont avantageusement remplacés par des chassis en plastique. Les sacs en plastique sont dans tous les magasins (et aussi dans les océans ou les forêts) et peuvent supporter des charges lourdes sans problème (quand ils cassent c’est généralement en raison d’une arête de coupe). Le développement des polymères était vraiment une révolution dans nos vies et nous pouvons difficilement imaginer un retour en arrière.
Les polymères sont dérivés du pétrole et sont faits de petits blocs appelés monomères. Les monomères sont de petites molécules, généralement gazeuses, portant un groupe fonctionnel qui peut donner des propriétés au polymère. Les monomères, qui composent un polymère, sont ajoutés un par un au polymère qui se développe en longueur. Nous verrons la réaction un peu plus tard. En raison de leur taille énorme les polymères sont solides. Pour écrire la formule d’un polymère nous écrivons le plus petit bloc constitutif entre crochets, ce bloc étant présents « n » fois dans le polymère. A noter que pour un réacteur les longueurs des polymères produits ne sont pas toutes identiques. Nous obtenons une distribution de taille et « n » n’est donc pas une valeur fixe.
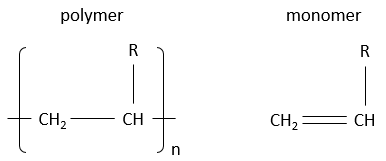
Voici quelques-uns des polymères habituels et les monomères dont ils sont faits.

Les propriétés d’un matériau polymère sont données par les interactions entre les chaînes de polymère. Les chaînes peuvent être linéaires ou ramifiées. Il est possible de fabriquer des polymères de plusieurs monomères différents. Il ya plusieurs façons d’obtenir ces polymères. Les monomères dans la chaîne peuvent être mis en bloques, en series ……..

Comme écrit plus haut les propriétés des polymères dépendent des interactions entre les chaînes. S’il n’y a pas d’interaction du tout les chaînes se déplacent librement entre elles, sans résistance et si nous étendons le matériau par deux côtés opposés il se dépense et se brise. La même chose arrive si les chaînes ont des attractions physiques entre elles. Celles-ci peuvent facilement être cassées par un chauffage ou par la force. Ce genre de matériel est appelé thermoplastique.
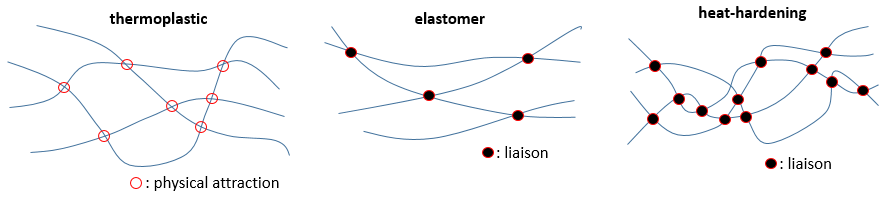
Des liaisons peuvent être faites entre les chaînes. S’il n’y en a que quelques-unes lorsque nous tendons la matière celle-ci va s’étendre jusqu’à un point où les liaisons bloquent tout mouvement supplémentaire. Elles empêchent le matériel de se briser aussi facilement qu’un produit thermoplastique. Un tel produit est appelé un élastomère.
S’il y a plus de liaisons le produit devient plus rigide et la rigidité augmente lorsque nous le réchauffons celui-ci est appelé un produit thermo-durcissant.
Courbe de traction-tension :
L’élasticité et la rigidité des polymères sont déterminées par une courbe de traction. Une machine étire le matériel alors que la force appliquée et l’étirement (ou la déformation) de la matière sont mesurés. Le matériel doit avoir la géométrie d’un cylindre pour éviter les problèmes liés à la symétrie. Au lieu de la force on mesure la pression (ou tension) R = F / S où S est la surface de la base du cylindre.
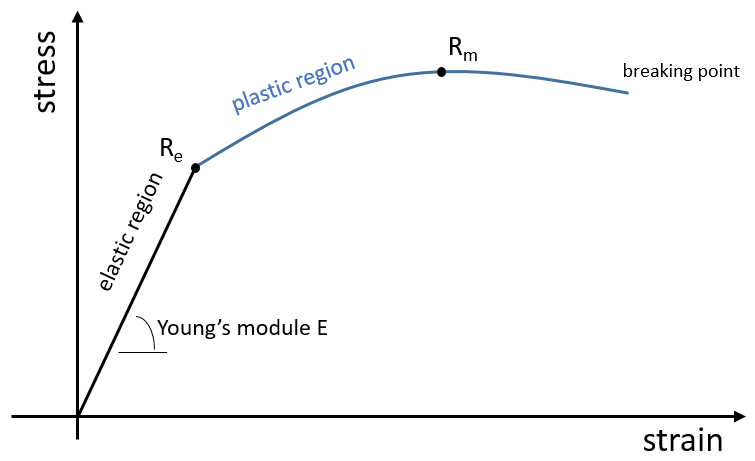
Dans un premier temps la déformation est élastique et la variation de la longueur de l’échantillon ΔL est directement proportionnelle à la force appliquée F. La pente de cette courbe donne le module de Young E qui est une caractéristique de l’élasticité (dans la direction donnée) de la matière. Si la force devait être enlevée le matériel serait revenu à sa forme initiale. C’est donc un processus réversible.
Ensuite la courbe s’aplatit et la déformation n’est plus une déformation élastique mais une déformation plastique : le matériel est déformé par la force appliquée et ne peut pas revenir à l’état initial. La variation de la pente peut être légère ou massive. Cette région est appelée la région d’écrouissage et le point où le changement de pente survient est appelé la limite d’élasticité Re. La courbe devrait augmenter jusqu’à un maximum Rm appelé la résistance à la traction maximum. Pourtant tous les nœuds ne se cassent pas toujours en même temps et nous pouvons voir l’apparition des plateaux sur la courbe. Au Rm les liaisons liant les polymères sont toutes cassées. L’expérience se poursuit jusqu’à ce que le matériel soit cassé en deux morceaux distincts.
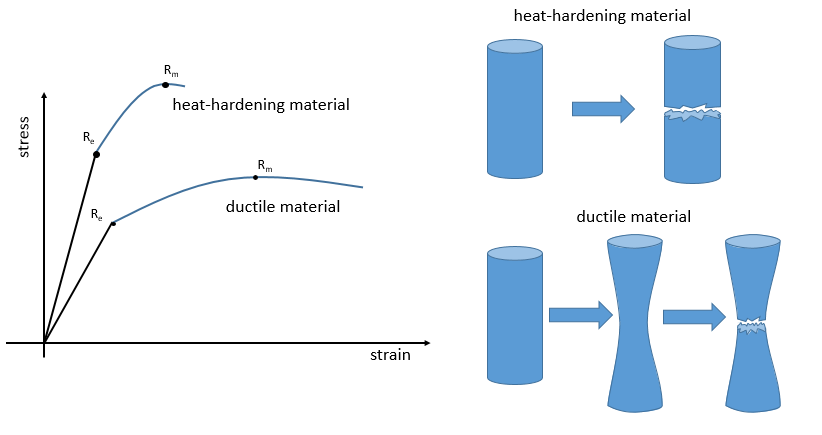
La ductilité désigne la façon dont un matériau se rompt. Les matériaux thermodurcissables se cassent après une région élastique courte (avec un grand module E de Young) : à cause de beaucoup de liaisons le matériel n’ est pas flexible mais quand le stress est assez grand, le matériel casse directement en deux morceaux. Dans le cas de matériaux ductiles l’échantillon devient plus mince et plus long avant la fracture. En conséquence la courbe continue plus loin après Rm.
Les paramètres qui influent sur les propriétés des polymères sont :
a) La température : par les interactions entre les chaînes. À un certain point appelé la température de transition vitreuse TG, les propriétés du polymère changent de manière drastique. La température de transition vitreuse TG est un intervalle de caractéristique de température, pour un matériau donné, de la transition entre l’état solide et un état flexible et élastique. TG dépend de tous les paramètres suivants et de la pression.
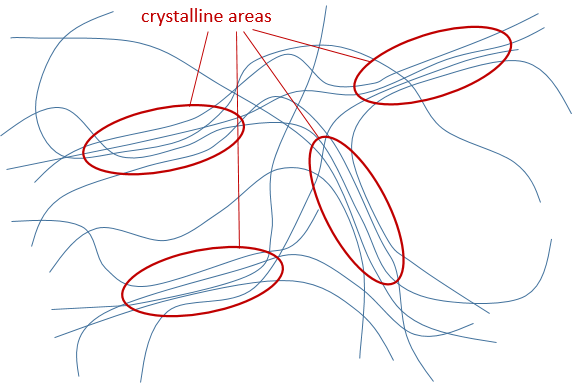
b) La cristallinité : il se réfère à l’ordre dans le solide. Les interactions et la densité dans le polymère ne sont pas identiques partout dans le matériau. Les zones cristallines sont des zones où les chaînes sont bien emballées et ont des interactions fortes. Les chaînes dans les zones amorphes ne sont pas bien emballées. Une chaîne unique peut être en même temps dans une région amorphe et dans une zone cristalline.
c) La longueur de la chaîne : il augmente la quantité d’interactions.
d) Le monomère(s) : le groupe que le monomère porte influence des interactions entre les chaînes
e) La réticulation : c’est la quantité de liaisons chimiques que nous faisons dans le matériel.
f) la vitesse de déformation : un matériel élastique peut se briser si nous tendons assez vite.
g) Les additifs : nous pouvons ajouter certaines molécules dans le matériel pour lier les chaînes ensemble ou pour éviter que cela se produise.
La synthèse :
Il ya plusieurs façons de faire des polymères. On peut les subdiviser en deux types : la polyaddition et polycondensation. La différence est que la polyaddition génère seulement le polymère tandis que la polycondensation génère également des petites molécules.
Polyaddition :
Elle peut être subdivisée en deux grandes catégories : la polymérisation radicale (polymérisation non chargée) et la polymérisation ionique (anionique, cationique ou polymérisation de coordination). Ils sont tous semblables mais nous n’allons montrer que la polymérisation radicale ici.
La synthèse est composée de 3 étapes principales : l’initiation, la propagation et l’étape de terminaison.
L’étape d’initiation :
Un initiateur est mélangé avec les monomères. Dans le cas de la polymérisation radicale l’initiateur est un radical. Ce radical peut être généré par la décomposition thermique ou par photolyse.
Par exemple le peroxyde de benzoyle (BPO) peut être clivé de manière homolytique par une élévation de température ou par un laser d’une fréquence donnée.
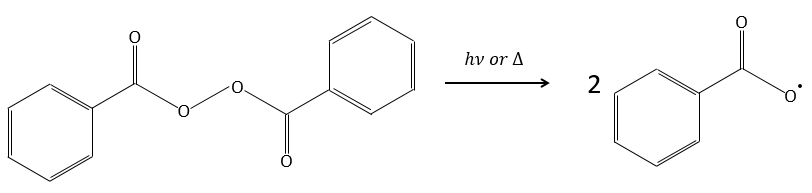
En fait le radical produit se réarrange pour libérer le CO2.
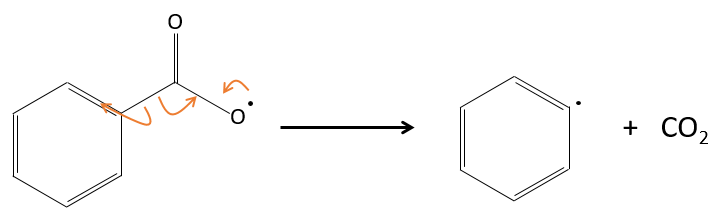
Les radicaux réagissent avec un monomère, le transfert du radical de l’autre côté de la molécule.

De ce point nous avons un début de chaîne qui porte un radical et qui est capable d’attaquer les monomères c’est l’étape de propagation :
![]()
Comme il existe encore un radical à la fin de cette réaction il va ainsi de suite, ce qui augmente la longueur de la chaîne par un monomère à chaque fois que la réaction a lieu. Une chaîne arrête de croître lorsque le radical est neutralisé. C’est l’étape de terminaison. Lors de cette étape deux chaînes en croissance réagissent ensemble. Cela peut arriver de deux façons :
Résiliation (terminaison) : les deux chaînes se confondent
![]()
Dismutation : les extrémités des chaînes portant le radical ont une réaction d’oxydoréduction ensemble. Pour rappel la réaction de dismutation d’habitude est un type spécifique de réaction redox dans laquelle une espèce est simultanément réduite et oxydée pour former deux produits différents.
![]()
Dans le cas des polymères :
![]()
Polycondensation :
Le processus est tout à fait différent de la polyaddition. Dans ce cas les monomères portent un groupe fonctionnel à ses deux extrémités. Les groupes fonctionnels peuvent réagir avec ceux des autres monomères à condenser ensemble. Par exemple un acide carboxylique se condense avec un alcool, un ester et l’eau.
![]()
Si l’alcool a été remplacé par un groupe amine nous obtiendrions un amide qui est aussi d’habitude la liaison entre les peptides et donc appelé une liaison peptidique.
![]()
La production de polymères est un processus exothermique : nous créons de nouvelles obligations. Toutefois il diminue le trouble comme il y a moins de molécules dans le système. En conséquence la production de polymères doit être effectuée dans une plage donnée de température. La température peut être approchée à partir du type et de la concentration des monomères. En effet, sauf au début de la réaction, il faut que la concentration de deux polymères de tailles similaires soient approximativement égales.
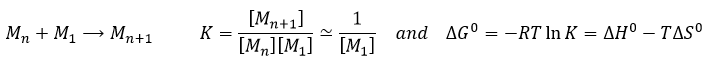
Chapitre 5: Cinétique chimique – vitesse de la réaction
Cinétique chimique :
La cinétique est un domaine de la chimie qui étudie la vitesse des réactions. La vitesse de la réaction peut dépendre des conditions de la réaction. Par exemple lorsque nous mettons H2 (g) et O2 (g) nous ne produisons pas de l’eau spontanément :
![]()
Cette réaction a une ΔG0 très négative. Cependant si nous produisons une étincelle la réaction va aller très vite.
Nous avons vu que la température a une influence sur l’équilibre des réactions. Au SCTP la formation d’ammoniac a une ΔG0 négative mais devient positive pour des températures de plus de 434 K :
![]()
Sans variation de température une réaction peut être accélérée si l’on utilise un catalyseur. Nous allons discuter des catalyseurs plus loin dans ce chapitre.
Il peut donc être important de connaître la vitesse des réactions en fonction de différents paramètres afin d’optimiser la production d’une molécule particulière, la destruction de certains déchets ou tout autre procédé.
La vitesse d’une réaction est une variation de la concentration des espèces engagées dans la réaction en fonction du temps. Elle est toujours positive. Comme il peut y avoir plusieurs différents coefficients stoechiométriques, nous devons nous entendre sur la définition de la vitesse d’une réaction parce que les concentrations ne varient pas de la même façon. Pour obtenir cette vitesse, on divise la variation dans le temps de la concentration des espèces correspondantes par le coefficient stoechiométrique. Dans la réaction :
![]()
nous considérons une seule vitesse de réaction qui est :
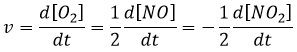
Les concentrations des réactifs diminuent au fil du temps et le débit est de signe opposé à la variation de leur concentration :
Pendant la réaction la vitesse dépend de la concentration de toutes les espèces impliquées :
![]()
Les coefficients α, β et γ sont l’ordre de la réaction en ce qui concerne les espèces correspondantes et leur somme α + β + γ est l’ordre global de la réaction. L’ordre de la réaction à l’égard de l’une des espèces est souvent leur coefficient stoechiométrique mais pas systématiquement.
A l’équilibre la vitesse de la réaction dans une direction est égale à la vitesse de la réaction dans l’autre sens :
![]()
Il est faux de dire que v = 0. Les deux réactions ont la même vitesse.
Au début de la réaction on peut négliger les concentrations des produits de l’expression de la vitesse avec β = γ = 0. La vitesse au début de la réaction est donc simplement :
![]()
Méthode intégrale :
On peut déterminer l’ordre de la réaction en ce qui concerne une espèce en gardant constante la concentration des autres réactifs. Fondamentalement nous mettons un grand excès de réactifs sauf pour les espèces que nous voulons étudier. De cette façon, seulement, une concentration varie de façon significative. Pendant la réaction on détecte la variation de la concentration de l’espèce cible dans le temps :
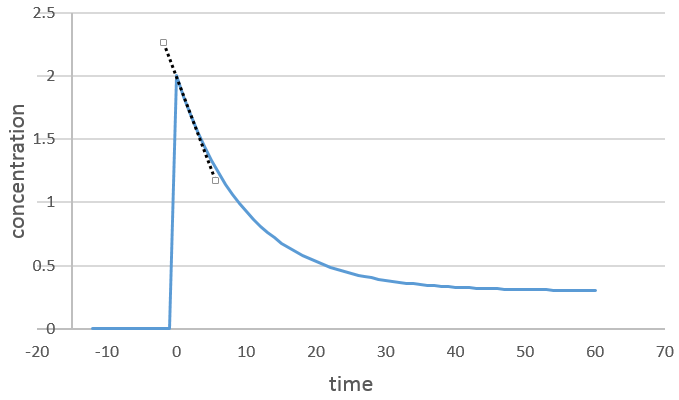
Nous nous concentrons sur la période de temps juste après le début de la réaction. Avant cela il n’y a aucune variation détectable (le réactif n’est probablement même pas dans la solution), puis les produits peuvent jouer un rôle dans la vitesse de réaction.
Si la réaction a un ordre égal à 1 à l’égard des espèces cibles la vitesse est :
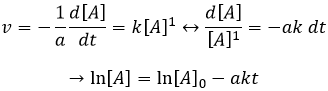
Et ainsi nous pouvons trouver une ligne droite lorsque nous traçons ln [A] en fonction du temps dont la pente est -ak :
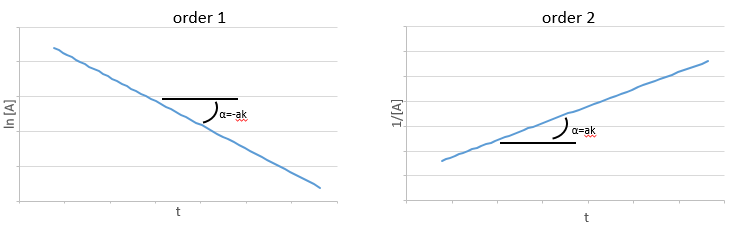
Si l’ordre = 2, le ln [A] ne donnera pas une ligne droite mais 1/[A] le fera. Dans ce cas la pente est ak :
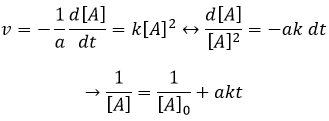
L’ordre peut également être égal à zéro ce qui signifie que la concentration du réactif n’a pas d’importance (sauf pour de très faibles concentrations). Cela signifie que la réaction est limitée par quelque chose d’autre. Nous observons souvent un ordre zéro avec les réactions qui se déroulent en présence d’un catalyseur.
Les catalyseurs sont des espèces qui affectent la réaction mais ne sont pas consommés pendant la réaction. Les catalyseurs les plus communs sont solides mais certains catalyseurs sont liquides. Sur les catalyseurs solides les réactifs peuvent se lier. En raison de cette nouvelle liaison, les liaisons dans le réactif sont plus faibles et la réactivité est améliorée. La réaction est ainsi limitée par l’espace disponible sur le catalyseur. La solution est mélangée au cours de la réaction afin qu’il y ait toujours des réactifs au voisinage du catalyseur. Sinon les réactifs seront de plus en plus loin du catalyseur au fur et à mesure que le temps passe. L’évolution de la concentration serait plus complexe dans ce cas. Nous allons discuter à propos des catalyseurs plus tard.
La méthode des vitesses initiales :
Au lieu d’utiliser des excès de réactifs nous pouvons faire plusieurs expériences et varier la concentration, à chaque fois :
![]()
n’est évidemment pas une réaction simple. La vitesse dépend simultanément des trois réactifs BrO3–, Br– et H+. Nous faisons d’abord la réaction avec des concentrations identiques [BrO3–] = [Br–] = [ H+] = 0.1M. Nous analysons l’évolution de la concentration d’une espèce en fonction du temps et on ne considère que la variation de concentration directement après le début de la réaction pour déterminer la vitesse de la réaction :
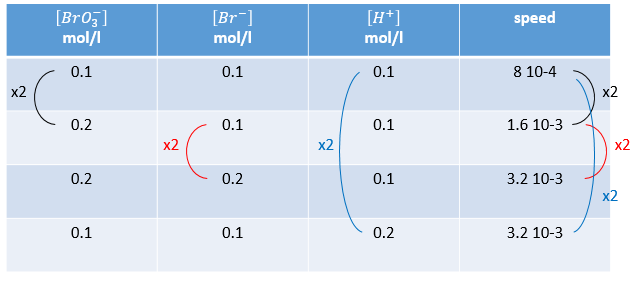
Dans un second temps nous faisons la même réaction mais modifions la concentration d’un des réactifs. La vitesse de réaction va changer en conséquence et ainsi nous pouvons déterminer l’ordre pour chaque espèce. Finalement lorsque nous savons tous les ordres nous pouvons déterminer la valeur de k.
Modèle de collisions :
Les réactions sont décomposées dans les événements élémentaires. Dans ce cas l’ordre est le coefficient stoechiométrique (généralement 1 ou 2). Les événements élémentaires sont des réactions qui sont effectuées en une seule étape : tout est fait lors de la collision entre les deux molécules :
![]()
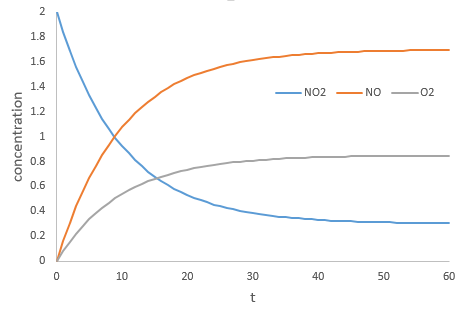
Par exemple la réaction entre le monoxyde de carbone CO et le NO2 est un événement primaire : l’atome d’oxygène est transféré de NO2 au CO lors d’une collision entre les deux espèces. Lorsque l’impact se produit et les molécules sont dans la bonne orientation la liaison N-O s’affaiblit et simultanément une liaison est formée entre C et O :
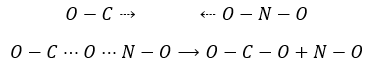
Les collisions entre les molécules ne conduisent pas toujours à un nouveau produit et il y a des conditions pour que la réaction se produise. Le produit intermédiaire de réaction OCONO ne peut être obtenu que si la collision vient d’un angle donné : l’intermédiaire est linéaire. En plus de cette limitation spatiale pour transférer l’atome d’oxygène la liaison entre O et N doit être étirée, ce qui nécessite de l’énergie. Enfin il existe une variation du nombre de moles de gaz au cours de cette réaction : NO est un liquide et par conséquent il implique une diminution de l’entropie qui doit être entièrement dégagée.
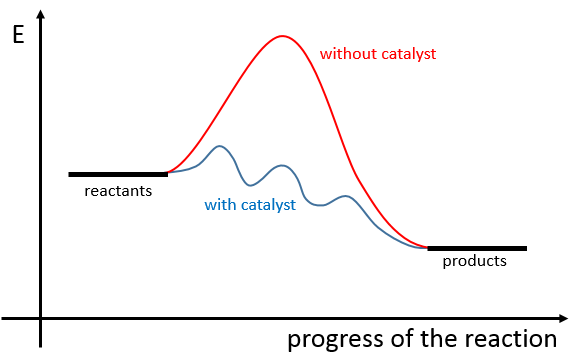
Il existe donc un minimum d’énergie qui est nécessaire pour obtenir la réaction. Cette énergie est appelée l’énergie d’activation. On peut représenter l’évolution d’une réaction par le graphique suivant :
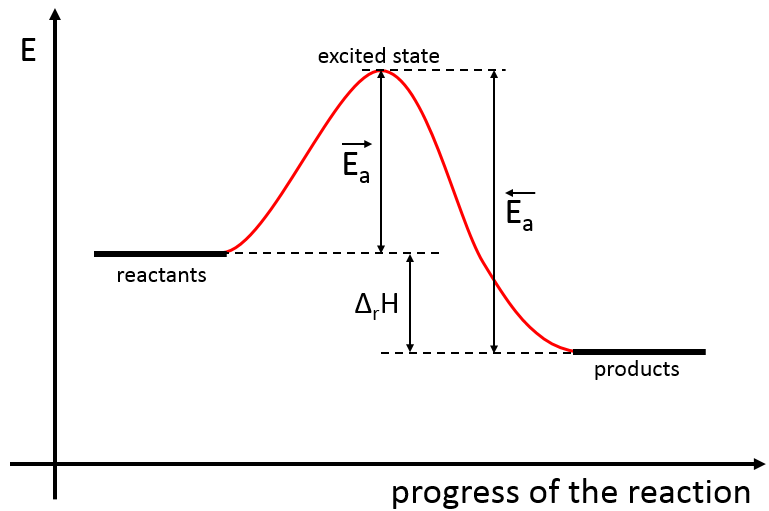
Les réactifs, pris séparément, ont une quantité donnée d’énergie. Les produits de la réaction ont une énergie plus faible. La différence d’énergie entre les produits et les réactifs est l’enthalpie de la réaction. Pour obtenir les produits l’énergie d’activation est similaire à une barrière qui doit être montée. Au sommet de cette barrière se trouve l’état excité, appelé de cette façon parce qu’il est riche en énergie. Après l’état excité l’énergie diminue vers le bas pour atteindre le niveau d’énergie, des produits, qui doit être inférieur à l’énergie des réactifs.
Pour faire la réaction dans l’autre direction, en formant du CO et NO2, l’énergie d’activation est beaucoup plus grande et les produits sont énergétiquement moins favorables. La différence entre les deux énergies d’activation est aussi égale à l’enthalpie de réaction.
Pour dépasser l’énergie d’activation les molécules doivent avoir assez d’énergie cinétique. Si tel est le cas elles auront des collisions avec succès. Rappelez-vous que l’énergie cinétique des molécules dépend de la température. Si la température augmente une plus grande population de molécules aura une énergie cinétique supérieure à l’énergie d’activation.
Dans l’expression de la vitesse de la réaction on trouve ces dépendances dans la constante de vitesse k :
![]()
Le paramètre d’énergie se trouve dans l’exponentielle et le paramètre spatial et la fréquence, des collisions, se trouvent dans la constante A.
Notez que les molécules lentes peuvent également réagir si elles ont une grande énergie potentielle par exemple beaucoup d’énergie de vibration. Au lieu de l’énergie cinétique l’énergie potentielle est utilisée :
![]()
Par conséquent si deux NOBr sont correctement alignés ils peuvent former Br2 même si leur énergie cinétique est faible en raison de la vibration des liaisons Br-N :
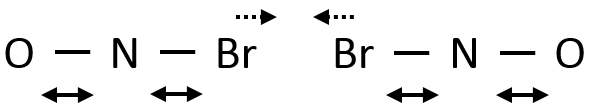
Eventuellement les liaisons N-Br peuvent être étirées simultanément, en approchant les deux Br l’une de l’autre on peut faciliter la formation de Br2.
La vitesse de la réaction pour former l’acide iodhydrique à partir d’hydrogène et d’iode peut nous faire penser que c’est une réaction élémentaire :
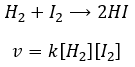
En effet les exposants des concentrations sont les coefficients stoechiométriques des réactifs. Toutefois cette réaction n’est pas une réaction élémentaire. Pour être une réaction élémentaire il faudrait que les molécules soient alignées d’une manière spécifique et que les liaisons se cassent et se forment en même temps. Cela signifie que les quatre événements doivent se produire en même temps, en plus de l’angle de collision spécifique :
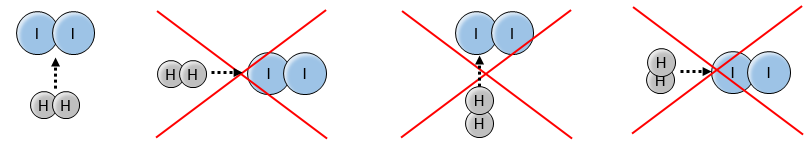
Pourtant la réaction se fait à une vitesse telle que nous pouvons penser à un événement élémentaire. Alors que la réaction complète est la succession de plusieurs évènements élémentaires qui se produisent l’un après l’autre.
La première étape est la dissociation homolytique de I2. Cela se fait à l’aide d’un métal catalyseur :
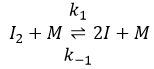
Il a été observé que la composition du récipient peut affecter la vitesse de la réaction. La dissociation se fait donc à la surface du récipient. Après la dissociation I peux réagir avec H2 pour former H2I qui peut réagir avec le second I de la première réaction pour produire 2 HI.
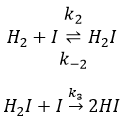
Alors que la dernière réaction est complète, la première et deuxième réaction sont des réactions d’équilibre et peuvent donc aller dans les deux sens.
La vitesse globale de la réaction est déterminé par l’événement primaire le plus lent. Cette étape lente est la dernière réaction :
![]()
Comment nous pouvons prouver que cette vitesse est égale à celle obtenue expérimentalement?
![]()
Pour obtenir une telle chose, nous utilisons deux hypothèses :
les réactions d’équilibre sont presque à l’équilibre : la troisième réaction est plus lente que les deux premières réactions et on suppose que la différence de vitesse est telle que les premières réactions (presque) ont le temps pour atteindre l’équilibre. Par conséquent, on peut dire que pour ces deux réactions la vitesse dans une direction est égale à la vitesse dans la direction opposé :
![]()
Le rapport entre les constantes de réaction k1 / k-1 est la constante K1 global.
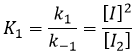
Nous pouvons faire la même chose pour la deuxième équation :
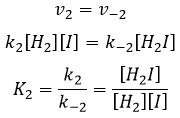
Nous pouvons maintenant remplacer les concentrations de v3 avec les expressions que nous avons trouvées tout à l’heure :
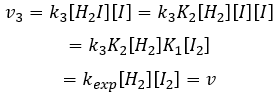
La constante qui a été trouvée expérimentalement est donc une combinaison des trois constantes de réaction.
la concentration de H2I est constante :
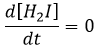
H2I est le produit de la deuxième réaction et le réactif de la troisième réaction. Par cette hypothèse nous supposons que dès que H2I est produit, il sera consommé par l’étape suivante de la réaction ou redevient H2 et I par l’inverse de la deuxième réaction.
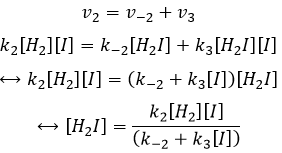
Nous pouvons insérer cela dans l’expression de la v3 :
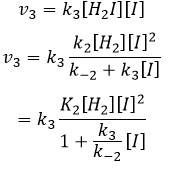
Étant donné que v1=v-1 (à partir de la première hypothèse), [I]2=K1[I2] et :
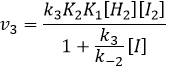
Nous avons ici la même expression que celle que nous avons obtenue avec la première hypothèse dans le numérateur. Nous pouvons donc faire l’hypothèse que la concentration de H2I est constante si K3 [I] / k-2 << 1, à savoir que la concentration de I est petite et que k3<<k-2. Elle est donc seulement correcte au début de la réaction.
Nous pourrions penser que l’hydrogénation de Br2 suit la même cinétique, mais nous sommes loin de la vérité. En effet :
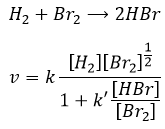
Cette réaction est également une série d’étapes élémentaires
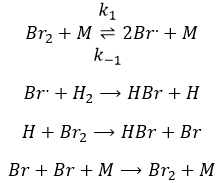
Le catalyseur :
Un catalyseur est une espèce qui est impliquée dans une réaction mais non consommée ou produite pendant la réaction. Il permet à la réaction de se produire par une diminution de l’énergie d’activation.
Un catalyseur peut être dans la même phase que les réactifs, dans ce cas on parle d’un catalyseur homogène ou dans une phase différente, comme par la réaction entre H2 et I2 dans lequel le catalyseur est la surface du récipient, un solide. Dans ce cas nous parlons d’un catalyseur hétérogène. Il existe au moins 4 étapes de l’action du catalyseur. Nous allons les voir dans la réaction de l’hydrogénation catalytique de l’éthylène :
Adsorption et activation des réactif s: les espèces qui se trouvent dans le voisinage du catalyseur hétérogène peuvent se lier à lui. En terme d’entropie la liaison est assez neutre : les espèces proviennent d’un liquide à lier sur une surface où beaucoup de places sont possibles. Il existe donc un grand nombre de configurations possibles sur la surface.
Migration des réactifs sur la surface : les réactifs doivent être à proximité les uns des autres pour réagir ensemble
Réactions
La désorption des produits
Dans le cas d’un catalyseur homogène le catalyseur est juste une espèce qui intervient au cours de la réaction mais non consommé par elle. Dans des niveaux élevés de l’atmosphère, le NO agit comme un catalyseur dans la destruction de l’ozone.
Le cas de la couche d’ozone :
La formule de l’ozone est O3. Cette molécule est sensible aux rayons UV qui la divise en O2 et un radical O.. Un radical est une espèce ou une molécule qui a des électrons de valence non appariés et qui est très réactif. Il est généralement le résultat d’une dissociation homolytique, à savoir les électrons de la liaison sont partagés de manière égale entre les deux atomes. Il est si réactif qu’il peut briser des molécules voisines pour former une liaison. Nous utilisons des radicaux pour produire des polymères (qui seront vus dans le chapitre correspondant) et aussi dans les produits antibactériens. Le bon point de l’utilisation de l’ozone comme un produit antibactérien est qu’il rejette l’oxygène O2 en tant que déchet, de sorte qu’il est propre pour l’environnement.
La destruction de l’ozone se fait en deux étapes : la première étape est la dissociation d’un radical O. sous le rayonnant de photons UV.
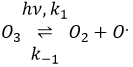
Le radical peut réagir avec l’O2 (réaction inverse) pour revenir à l’ozone ou de réagir avec une autre molécule d’ozone pour former plus d’O2.
![]()
Au total on obtient 3 O2 à partir de 2 molécules O3.
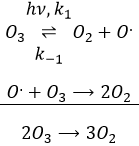
Le radical O. n’est pas un catalyseur de la réaction même si nous ne trouvons pas dans la réaction globale car elle est produite et consommée pendant la réaction. C’est une espèce qui est déjà présente dans le système dès le début et qui reste dans le système une fois que la réaction est terminée. La vitesse que nous trouvons expérimentalement est :
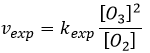
Ce n’est pas la vitesse que nous pourrions nous attendre d’une réaction en une seule étape. Nous pouvons faire l’analyse cinétique de la réaction. L’étape de détermination de cette réaction est la deuxième réaction.
![]()
La concentration du radical est difficile à déterminer mais nous pouvons encore faire les mêmes hypothèses que nous avons faites avec la réaction entre H2 et I2 : la concentration du produit intermédiaire ne change pas et la première réaction est à l’équilibre :
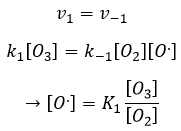
On peut insérer cela dans l’expression de la vitesse :
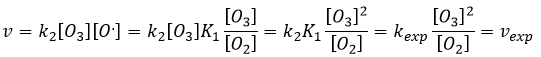
Freon’s
Freon’s are small species full of halogens, CCl2F2 for instance that have a very high heat capacity and are nonpolar and inert. There have been used as refrigerant in the fridges for a long time and we thought that, as those species were inert in the labs we had no specific care to apply to them. However Freon’s are deteriorated by UV rays. In dumps, the tubes containing the Freon eventually break and the gas goes up high in the atmosphere where it is not inert anymore. The deterioration of Freon’s due to UV’s leads to the formation of radicals that will interact with the ozone.
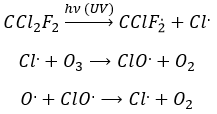
If we sum the two last equations, we get the determining step of the deterioration of the ozone alone.
![]()
The Cl. is thus a catalyst of the destruction of the ozone and the hole in the ozone layer started to grow. The problem is that the Freon gas takes a very long time (years) to reach the higher atmosphere and at the moment we observed their effect, a lot of Freon’s were already climbing. Now the use of Freon’s is forbidden and the hole in the ozone layer is slowly decreasing in size.
Another product having an effect on the ozone is some nitrogen products of the exhaust pipes of cars. During the combustion of the fuel, nitrogen monoxide and dioxide can be formed.
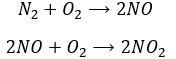
Nitrogen dioxide is deteriorated by light.
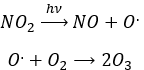
Low in the atmosphere, and at the ground level, NO acts as a catalyst and generates peaks of ozone. Higher, NO catalyses the destruction of the ozone.
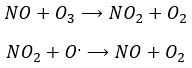
Chapitre 4: Cinétique chimique – Équilibre
La cinétique chimique est l’étude de la vitesse des réactions. La vitesse de réaction est déterminée par la variation de la quantité d’espèces par unité de temps. Habituellement nous considérons les concentrations pour les espèces en solution et nous considérons les pressions pour les gaz.
La vitesse d’une réaction est rarement constante et positive jusqu’à ce que le système atteigne l’équilibre. A l’équilibre la vitesse de réaction est égale à zéro.
L’ équilibre :
imaginons la réaction suivante :
![]()
L’équilibre est atteint lorsque les concentrations des espèces ne varient plus dans le temps. La constante d’équilibre, qui exprime la loi d’action de masse, est :
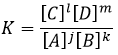
La constante de la réaction inverse (de droite à gauche) est K ‘= 1 / K et si l’on considère un multiple n de cette réaction on aura :
![]()
K »= Kn parce que les coefficients stoechiométriques sont les exposants de l’expression de la constante d’équilibre.
Les unités de K dépendent de la réaction. Par exemple la réaction de Haber qui génère de l’ammoniac et que nous avons déjà rencontré avant :
![]()
a une constante d’équilibre Kp qui dépend de la pression des réactifs et des produits.
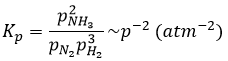
Les unités de K dépendent donc de la quantité des différents produits et réactifs et de leurs coefficients stoechiométriques. Nous notons la constante avec un P parce que nous considérons habituellement les constantes d’équilibre en terme de concentrations que nous pouvons désigner Kc. Il y a une relation entre Kc et Kp que nous trouvons avec la loi des gaz parfaits PV = nRT
![]()
La relation entre la pression et la concentration est ainsi simplement une multiplication par RT. Dans l’expression de K ce RT est également à la puissance du coefficient stoechiométrique.
Pour la réaction de Haber nous trouvons :
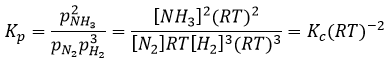
Dans le cas des phases condensées nous ne tiendrons pas compte de la concentration ni de la pression parce que les interactions entre les espèces doivent être prises en compte. Nous considérerons l’activité des espèces à la place de ces paramètres. L’activité est proportionnelle à la concentration d’une espèce ou à sa pression multipliée par un coefficient γ. L’équilibre peut être hétérogène si toutes les espèces qui interviennent dans la réaction ne sont pas de la même phase. Dans ce cas aussi nous considérerons les activités de l’espèce à la place de leur concentration ou de leur pression. Par exemple la réaction :
![]()
est une réaction où deux solides et un gaz coexistent. Il a la constante d’équilibre suivante :
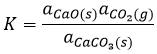
Nous choisissons volontairement une réaction impliquant des solides parce que leur activité est égale à 1. La constante est donc tout simplement proportionnelle à la pression de CO2. Dans un système fermé la réaction n’aura pas lieu même si nous augmentons la quantité de réactif. La réaction est seulement limitée par la pression du gaz.
Pour déterminer le sens d’une réaction nous comparons la constante d’équilibre K avec le coefficient réactif Q. Le Q est déterminé à partir des concentrations initiales mises en présence.
Pour la réaction :
![]()
le coefficient Q et la constante d’équilibre K sont respectivement
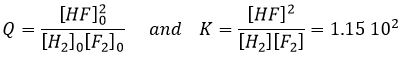
Si l’on considère un système de 5l contenant les quantités suivantes de chaque espèce :
![]()
Étant donné qu’il n’y a pas de produit dans cette composition la réaction ira évidemment de la gauche vers la droite. Il est impossible d’obtenir des concentrations négatives. La réaction va se poursuivre jusqu’à ce qu’elle atteigne l’équilibre ou que la totalité de l’un des réactifs soit consommée. On peut déterminer la concentration de chaque espèce à l’équilibre avec le procédé suivant :
Nous déterminons les concentrations des espèces. Ici nous avons donné le volume du récipient et le nombre de moles de chaque espèce. Nous les écrivons dans un tableau et supposons qu’une concentration x sera consommée pour atteindre l’équilibre. La même quantité (exprimée en rapports stoechiométriques) de produit a, sera formée :

Comme nous connaissons la valeur de K nous pouvons déterminer x et donc la concentration de chaque espèce.
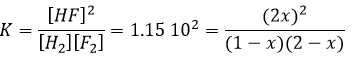
Cette équation du second degré donne deux solutions mais l’une est négative et donc impossible (concentration négative est impossible). Nous pouvons ainsi déterminer les concentrations à l’équilibre pour toutes les espèces.
Si K est faible, par exemple dans la réaction, cela signifie que la réaction ne consomme pas beaucoup de réactifs.
à K=1.6 10-5 mole/l. Si nous commençons la réaction avec seulement 1 mole de NOCl (g) nous allons consommer 0.03mole/l de celui-ci.

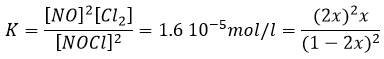
En supposant que x est faible la partie inférieure de la fraction est simplifiée et :
![]()
L’hypothèse selon laquelle 2x <<< 1 est correcte. Une petite constante d’équilibre signifie également que la réaction inverse est plus favorable (K ‘= 1/K).
Température d’inversion :
L’équilibre dépend de la température. Nous avons vu dans les sections de la thermodynamique que :
![]()
A une température donnée, les réactions vont dans un sens mais si nous augmentons la température suffisamment la réaction va aller dans la direction opposée. La température à laquelle l’inversion de la direction de la réaction se produit est la température d’inversion qui est égale à :
![]()
Si la réaction est effectuée à la p = 1 atm, nous pouvons trouver cette température à partir des tables de ΔH0 et ΔS0:
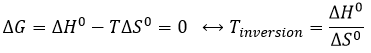
La modification de l’équilibre :
Maintenant si l’on considère un système à l’équilibre et que nous appliquons une modification sur ce système, Le Chatelier énonça que l’équilibre va changer pour contrebalancer cette modification.
– Ajout d’un substrat : l’équilibre va consommer plus de ce constituant ou produire moins de celui-ci.
Par exemple prenons une réaction à l’équilibre :
![]()
si nous ajoutons plus de A dans le système l’équilibre va se déplacer vers la droite pour consommer une partie du réactif ajouté :
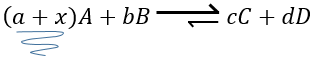
si on ajoutait une certaine C (un produit) l’équilibre serait déplacé vers la gauche :
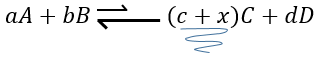
Une autre façon de modifier l’équilibre est d’éliminer les produits du système pour déplacer l’équilibre vers la droite.
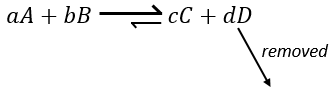
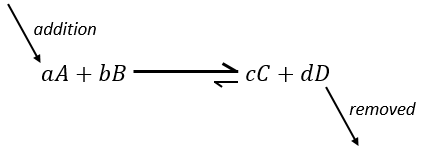
Pour optimiser la vitesse d’une réaction, on peut donc ajouter des réactifs et retirer les produits en permanence.
–Température : l’équilibre dépend de la température, comme nous l’avons vu ci-dessus. L’effet sera différent pour une réaction exothermique ou une réaction endothermique. En effet :
![]()
la variation de température affecte l’exponentielle avec ΔH0. Si ΔH0<0, soit il est une réaction exothermique, K diminue comme résultat d’une augmentation de la température et la réaction est donc moins efficace. Au cours d’une réaction exothermique la chaleur est un produit de la réaction :
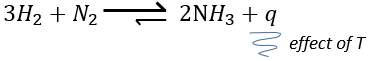
et la température augmente si la chaleur de l’équilibre est déplacé vers la gauche. Si la réaction est endothermique (la réaction inverse par exemple), une augmentation de température aide la réaction à se dérouler (plus de chaleur est disponible pour être absorbée). L’équilibre est donc déplacée vers la droite.
– pression/volume : au cours des réactions impliquant des gaz, une diminution du volume déplacera l’équilibre vers le côté où il y a moins de moles de gaz. Dans la réaction précédente l’équilibre sera déplacé vers la droite car on génère deux moles de gaz à partir de 4. On peut également déplacer l’équilibre par addition d’un gaz inerte qui augmentera la pression sans aucune modification de la réaction chimique.
Experimental determination of K, ΔH0 and ΔS0
On sait que l’équilibre est déplacé par une modification de la température. Imaginons que nous voulons connaître les valeurs de K, ΔH0 et ΔS0 d’un processus de dissolution comme :
![]()
on sait que s’il n’y a pas de Ag+ ou Cl– dans la solution avant la réaction leur concentration sera égale car durant la réaction chaque ion est produit de manière égale. Comme les solides ont une activité a = 1, la constante de dissociation est Ks :
![]()
La concentration en ions est donc égale à la racine carrée de la constante de dissociation. A une température donnée on peut donc mesurer la concentration à l’équilibre de l’un des ions pour déterminer la valeur de K. On peut faire cela à plusieurs températures et tracer la relation entre la température et K.
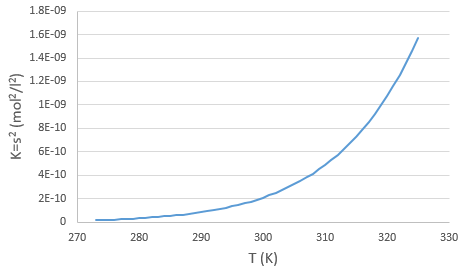
Nous voyons que la constante de dissociation K augmente avec la température ce qui signifie que, si nous augmentons la température, nous pouvons dissoudre plus de AgCl en solution. AgCl n’est pas très soluble dans l’eau : seulement …. mg par litre va se dissoudre (la racine carrée de Ks donne la concentration). Mais nous pouvons trouver plus d’informations à partir de cette relation. Nous connaissons déja l’équation reliant K et T :
![]()
Si nous traçons ln K en fonction de 1/T nous trouvons une ligne droite dont la pente est égale à ΔH0/R :
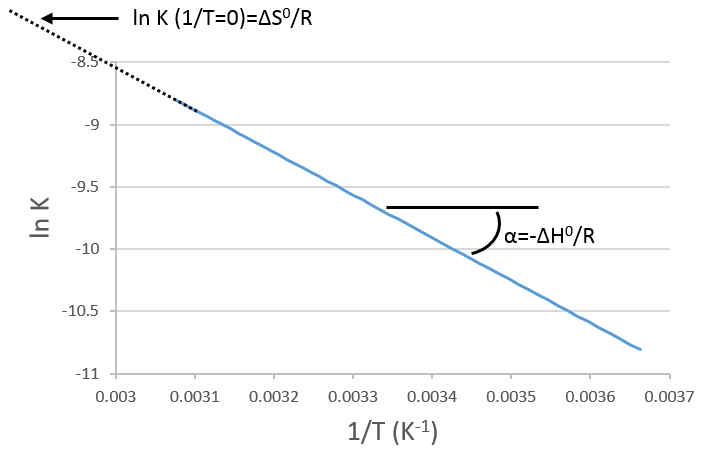
L’intersection avec l’axe à 1/T = 0 nous donnera ΔS0/R. Sur la figure l’axe n’ est pas à 1/T = 0 et l’intersection de la courbe avec l’axe ne signifie rien.
Chez le vivant ΔG est généralement très proche de zéro, de sorte qu’un stimulus peut permettre à une réaction d’avoir lieu. Dans notre corps, beaucoup de réactions, qui seraient normalement impossibles, fonctionnent car elles sont couplées avec l’élimination du phosphore d’un ATP pour permettre au processus d’avoir lieu. L’ATP est une réserve d’énergie de notre corps.
La cinétique chimique :
la cinétique est un domaine de la chimie qui étudie la vitesse des réactions. La vitesse de réaction peut dépendre des conditions de la réaction. Par exemple lorsque nous avons mis H2(g) et O2(g) nous ne produisons pas de l’eau spontanément :
![]()
Cette réaction a une ΔG0 très négatif. Cependant, si nous produisons une étincelle, la réaction va aller très vite.
Nous avons vu que la température a une influence sur l’équilibre des réactions. Au SCTP, la formation d’ammoniac a une ΔG0 négative mais elle devient positive à des températures de plus de 434 K.
![]()
Sans variation de température une réaction peut être accélérée si l’on utilise un catalyseur. Nous allons discuter des catalyseurs plus loin dans ce chapitre.
Il peut donc être important de connaître la vitesse des réactions en fonction de plusieurs paramètres afin d’optimiser la production d’une molécule particulière, la destruction de certains déchets ou tout autre procédé.
Vitesse de réaction :
La vitesse d’une réaction est une variation de la concentration des espèces engagées dans la réaction en fonction du temps. Elle est toujours positif. Comme il peut y avoir plusieurs coefficients stoechiométriques différents, nous devons nous entendre sur la définition de la vitesse d’une réaction parce que les concentrations ne varient pas de la même façon. Pour obtenir cette vitesse on divise la variation dans le temps de la concentration des espèces correspondantes par le coefficient stoechiométrique. Dans la réaction :
![]()
nous ne considérons qu’une seule vitesse de réaction qui est :
Les concentrations des réactifs diminuent au fil du temps et la vitesse est de signe opposé à la variation de leur concentrationb :
Pendant la réaction la vitesse dépend de la concentration de toutes les espèces impliquées.
![]()
les coefficients α, β et γ sont de l’ordre de la réaction en ce qui concerne les espèces correspondantes et leur somme α + β + γ est de l’ordre global de la réaction. L’ordre de la réaction à l’égard de l’une des espèces est souvent leur coefficient stoechiométrique mais pas systématiquement.
A l’équilibre la vitesse de la réaction dans une direction est égale à la vitesse de la réaction dans l’autre sens :
![]()
il est faux de dire que v = 0. Les deux réactions ont la même vitesse.
Au début de la réaction on peut négliger les concentrations des produits de la réaction dans l’expression de la vitesse car β = γ = 0. La vitesse au début de la réaction est donc simplement :
![]()
Méthode intégrale :
On peut déterminer l’ordre de la réaction en ce qui concerne une espèce en gardant constante la concentration des autres réactifs. Fondamentalement nous mettons les réactifs en excès, sauf pour les espèces que nous voulons étudier. De cette façon seulement une concentration varie de façon significative. Pendant la réaction on détecte la variation de la concentration de l’espèce cible dans le temps :
Nous nous concentrons sur la période de temps juste après le début de la réaction. Avant cela il n’y a pas de variation à détecter (le réactif n’est probablement même pas dans la solution) puis les produits peuvent jouer un rôle dans la vitesse de réaction.
Si la réaction est de l’ordre 1 à l’égard de l’espèce cible, la vitesse est :
et ainsi nous pouvons trouver une ligne droite lorsque nous traçons ln [A] en fonction du temps dont la pente est -ak :
Si l’ordre = 2, les ln [A] ne donnera pas une ligne droite, mais 1/[A] le fera. Dans ce cas la pente est ak :
La commande peut également être égal à zéro, ce qui signifie que la concentration du réactif n’a pas d’importance (sauf pour de très faibles concentrations). Cela signifie que la réaction est limitée par quelque chose d’autre. Nous observons souvent un ordre zéro réaction en présence d’un catalyseur.
Un catalyseur est une espèce qui affecte la réaction mais n’est pas consommé pendant la réaction. Les catalyseurs les plus communs sont des solides mais certains catalyseurs sont des liquides. Sur les catalyseurs solides les réactifs peuvent se lier. En raison de cette nouvelle liaison, les liaisons dans le réactif sont plus faibles et la réactivité est améliorée. La réaction est ainsi limitée par l’espace disponible sur le catalyseur. La solution est mélangée au cours de la réaction afin qu’il y ait toujours des réactifs au voisinage du catalyseur. Sinon, les réactifs, seront de plus en plus loin du catalyseur au fur et à mesure que le temps passe. L’évolution de la concentration serait plus complexe dans ce cas. Nous allons discuter plus longuement à propos des catalyseurs dans un autre chapitre.
Méthode des vitesses initiales :
Au lieu d’utiliser des excès de réactifs, nous pouvons faire plusieurs expériences et varier chaque fois la concentration d’une espèce. La réaction suivante :
![]()
n’est évidemment pas une simple réaction. La vitesse dépend simultanément des trois réactifs BrO3–, Br– et H+. Nous faisons d’abord la réaction avec des concentrations identiques [BrO3–] = [Br–] = [H+] = 0,1. On analyse l’évolution de la concentration d’une espèce en fonction du temps et on ne considère que la variation de la concentration directement après le début de la réaction pour déterminer une vitesse de la réaction.
Dans un second temps nous faisons la même réaction mais modifions la concentration d’un réactif. La vitesse de réaction va changer en conséquence et nous pouvons déterminer l’ordre pour chaque espèce. Finalement lorsque nous savons tous les ordres nous pouvons déterminer la valeur de k.
Chemical kinetics is the study of the speed of reactions. The speed of reaction is determined by the variation of the quantity of species by unit of time. Usually, we consider the concentrations for species in solution and we consider pressures for gases.
The speed of a reaction is rarely constant and is positive until the system reaches the equilibrium. At the equilibrium, the speed of reaction is zero.
Chapitre 3 : Méthodes de séparation – extraction
Des procédés de séparation :
Un des devoirs d’un chimiste est d’analyser des échantillons de produits chimiques. Pour déterminer la composition de l’échantillon, une séparation des différents éléments est nécessaire pour éviter les interférences entre les espèces.
Il peut être important de déterminer quels éléments chimiques sont présents, dans quelles quantités et dans quel état d’oxydation. Plusieurs méthodes de séparation peuvent satisfaire les besoins de la pharmacie pour obtenir une bonne séparation. Dans la plupart des cas l’échantillon doit être une solution dont le solvant ne contient pas d’espèces interférentes.
Les méthodes de séparation sont basées sur l’affinité des espèces, pour différents solvants, sur les processus d’équilibre et sur la cinétique. Certaines méthodes permettent d’obtenir l’échantillon de retour et certaines le détruisent.
Extraction :
Cette méthode est basée sur l’affinité de l’espèce avec deux phases liquides non miscibles différents. Une phase est aqueuse et la seconde est organique. L’expérience est réalisée dans une ampoule à décanter (ou entonnoir de séparation). C’est un morceau de verre ou de matière plastique en forme de cône renversé avec une vanne sur le fond et une ouverture à son sommet. Un bouchon peut fermer l’entonnoir pour permettre un mélange énergique.

L’échantillon est préparé dans une des deux phases et est placé dans l’entonnoir d’extraction par son ouverture supérieure. La deuxième phase est ajoutée (l’ordre d’insertion des deux phases n’a pas vraiment d’importance). Comme les phases ne sont pas miscibles, les échanges de particules entre les phases ne peuvent être faites à l’interface et des espèces chargées ne se déplacent pas dans la phase organique : leur solubilité dans toute phase organique est très faible. Pour améliorer les échanges les liquides sont mélangés en secouant l’entonnoir et en tournant à l’envers. Cela augmente la surface de contact entre les deux phases ainsi l’échange est amélioré. Au cours de la phase de mélange il est recommandé d’ouvrir brièvement l’entonnoir (par le capuchon ou par la valve) afin de libérer un éventuel excédent de pression. Rappelez-vous quelques notions de sécurité : il peut être nécessaire d’ouvrir l’entonnoir sous une hotte et ne pas mettre votre nez ou un oeil juste au-dessus de l’ouverture lorsque vous relâchez la pression. Ensuite le système est mis au repos jusqu’à ce que l’interface soit mince et claire. La phase de densité la plus faible se trouve en haut et la plus dense est au fond. Habituellement la phase organique au-dessus de la phase aqueuse en dessous car la densité des solvants organiques est généralement inférieure à la densité de l’eau (huile flotte sur l’eau). On peut ainsi éliminer la phase aqueuse hors de l’entonnoir par l’ouverture de la valve au fond de la fiole.
Répartition des espèces :
A l’équilibre chaque espèce est répartie entre les deux phases. On peut définir un coefficient de distribution KD qui est le rapport entre la concentration d’une espèce dans la phase organique et dans la phase aqueuse :

Si nous ne considérons pas spécifiquement l’équilibre c’est le rapport de distribution D.
![]()
Pour les concentrations de l’espèce nous considérons toutes les formes d’une espèce donnée. Certaines réactions peuvent se produire dans une phase mais pas dans l’autre et déplacer le rapport de distribution. Par exemple un acide dans la phase aqueuse peut être sous sa forme acide HA ou sous forme de base conjuguée A- mais la solubilité des ions dans les phases organiques est très faible.
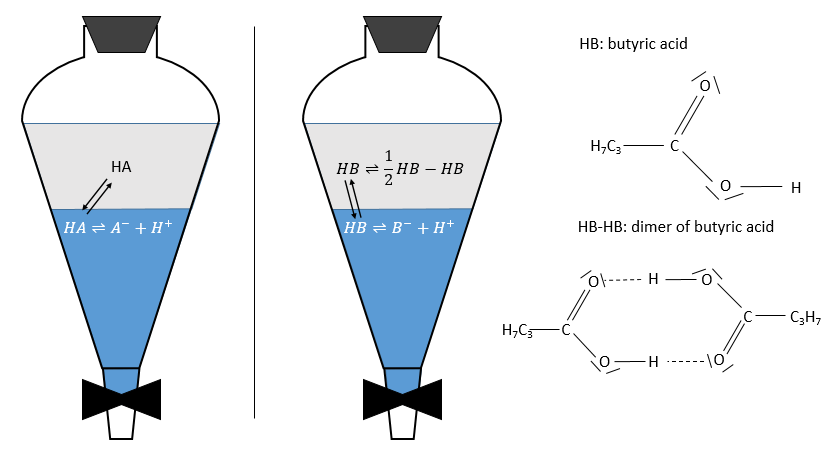
En conséquence les A- ne sont pas échangées et restent dans la phase aqueuse. D’autre part les espèces neutres HA ont une concentration équilibrée entre la phase aqueuse et la phase organique. Ainsi le rapport de distribution de l’acide est :
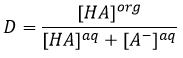
En conséquence le rapport de distribution peut dépendre de l’acidité de la phase aqueuse. Des équilibres peuvent également se produire dans la phase organique comme il est montré par l’exemple de l’acide butyrique. Dans la phase organique cet acide carboxylique peut former un dimère avec deux liaisons hydrogène. Dans ce cas le rapport de distribution sera
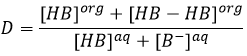
Le choix des solvants est donc crucial pour une bonne extraction. Les solvants peuvent être polaire ou non polaire avec la restriction qu’ils ne doivent pas être miscible.
Extraction des espèces organiques :
Pour extraire un composé organique de la phase organique, nous devons obtenir des espèces chargées. Les espèces neutres sont partagées entre les deux phases mais les ions restent dans la phase aqueuse. Si le composé n’est pas ionisable nous ne pouvons jouer que sur la nature du solvant pour avoir la meilleure extraction, à savoir lorsque KD est petit. Si le composé peut être dissocié on peut régler l’acidité de la solution aqueuse afin d’optimiser l’extraction.
L’acide acétique a un pKa de 4.7
![]()
A ce pH, dans la solution aqueuse, il y aura autant de CH3COOH que de CH3COO–. L’acétate peut être extrait dans la phase aqueuse. Si l’on utilise un acide plus fort la majorité de l’acide acétique qui pénètre dans la solution aqueuse va se dissocier pour former de l’acétate qui peut être extraite.
Un autre cas est le cas des espèces amphotères. Ces espèces peuvent jouer le rôle d’un donneur ou d’un accepteur de protons. L’oxime (8-hydroxyquinoléine) est un amphotère.

A pH <5 l’azote est protoné, donnant une charge positive au composé qui peut maintenant être extrait. A un pH> 10 le groupe hydroxyle perd son proton ce qui conduit à une espèce chargée négativement qui peut également être extraite.
Entre 5 <pH <10 oxime reste dans la phase organique.
L’extraction de métaux :
Les cations métalliques peuvent être extraits à partir de solutions aqueuses. Dans ce cas nous voulons obtenir une grande valeur de D. Pour transférer les métaux de la phase aqueuse leur charge doit être cachée par la formation de paires ioniques fortes ou par la formation de complexes de coordination. L’ oxime peut former un complexe avec des métaux pour les stabiliser dans la phase organique. Par exemple si l’on veut extraire l’aluminium d’une solution aqueuse, l’oxime peut se déplacer à partir de la phase organique à la phase aqueuse. Dans de bonnes conditions de pH, l’oxime perd son proton. Dans sa forme de base l’oxime forme un complexe avec les ions d’aluminium, un complexe qui peut maintenant être transféré dans la phase organique.
Fe3 + peut former une paire ionique dans une solution concentrée de HCl. La paire ionique (FeCl4) -H3O + peut se déplacer dans la phase organique. Notez que ce produit de la réaction se déplace hors de la solution aqueuse. L’équilibre est donc déplacé vers la droite.
Une autre façon de déplacer l’équilibre vers la droite est d’augmenter le pH de la solution aqueuse. Il est possible d’extraire plusieurs métaux spécifiquement en fonction du pH de la solution aqueuse. La figure suivante représente l’extraction de différents métaux par formation de dithizomates en fonction du pH.
On peut voir que l’extraction du plomb commence à pH> 6 tandis que l’Ag est déjà entièrement extrait à pH = 3. Pour la séparation du Cu (II) de l’Ag il faudra un autre procédé de séparation.
Fraction extraite :
Prenons l’extraction de l’acide butyrique. Le rapport de distribution D est le rapport de sa concentration dans la phase organique (éther) par rapport à sa concentration dans la phase aqueuse (eau). Après l’extraction 3g d’acide butyrique est dans la phase organique et 1g est encore dans la phase aqueuse.
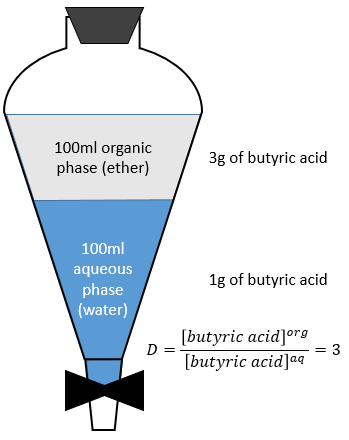
La fraction extraite F est la quantité de l’acide dans la phase organique sur la quantité totale de l’acide :

F = 3/4 si les volumes des phases aqueuses et organiques sont égales. Le rapport des volumes RV est le volume de la phase organique sur celui de la phase aqueuse. Si l’on considère un grand volume de solvant, à l’égard du volume de l’espèce à séparer, les volumes initiaux de solvants mis en présence peuvent être utilisés avec une bonne approximation. Après l’injection RV dans l’expression de F on obtient l’expression suivante :
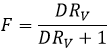
Nous pouvons conclure que l’extraction a de meilleurs résultats lorsque D ou RV est grande.
La fraction restante G est la fraction de l’échantillon qui n’a pas été transféré à la phase organique et est simplement
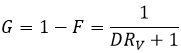
Maintenant si deux composés A et B doivent être séparés, nous devons concevoir l’expérience de telle manière que la multiplication de DA par DB soit égale à 1. En effet, si DA et DB sont grands mais DA >>> DB, la séparation ne sera pas efficace. Imaginez que DA = 1000 et DB = 20. (A) a une meilleure extraction que B mais les deux espèces sont extraites : les fractions extraites( en envisagent des volumes identiques de phases) seront :
![]()
Malgré l’énorme différence de D la séparation est mauvaise. Maintenant si DA = 10 et DB = 0,1 l’extraction de ces deux composés est plus faible que dans l’exemple précédent mais la séparation est bien meilleure :
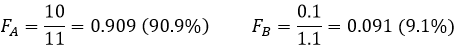
Cette fois-ci 91% de A sont dans la phase organique pour seulement 9% de B.
Pour obtenir une meilleure séparation nous pouvons répéter l’extraction à partir de la phase organique que nous venons d’extraire. Revenons à notre premier exemple de l’acide butyrique. Pour ce composé nous avions trouvé D = 3. Nous pouvons jouer sur les volumes de la phase pour modifier l’efficacité de l’extraction. Si nous mettons la moitié du volume de l’éther puis RV = 0,5 la fraction extraite sera :
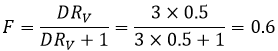
Sur les 4 premiers grammes 1,6 g sont dans la phase aqueuse et 2,4 g sont dans la phase organique. On pourrait avoir une meilleure extraction avec un plus grand volume d’éther. Cependant, si nous faisons une seconde extraction avec les mêmes conditions, F est à nouveau 0,6 et nous aurons 0,64 g d’acide butyrique restant dans la phase aqueuse.
Nous avons fait deux extractions avec 50 ml d’éther à la place d’une extraction avec 100 ml d’éther. Donc, avec le même volume, mais avec deux extractions, on a extrait plus de l’acide butyrique (3,36 g de 3 g vs). Plusieurs extractions avec un petit volume sont donc plus efficaces qu’ une extraction avec un volume important. Après n extraction, la fraction restante est

Appareil de Craig
Ce procédé a été optimisé dans l’appareil de Craig. Des dizaines à des centaines de tubes en forme de H sont connectés. La figure suivante montre un tube:
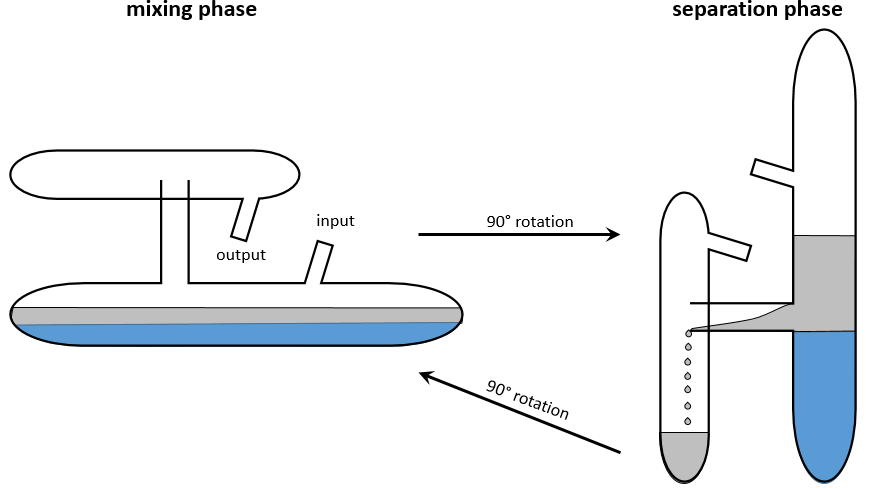
Les bras mécaniques mélangent les petits tubes au cours de la phase de mélange et les inclinent pour transférer la phase supérieure dans le petit compartiment du tube en forme de H. Une fois le transfert effectué les tubes sont remis dans leur position initiale. Cette rotation provoque la vidange du petit compartiment dans le tube suivant. Le processus est répété dans tous les tubes simultanément.
Dans un premier temps l’échantillon se trouve dans le premier tube (noté 0) et la phase inférieure est déjà dans tous les tubes :
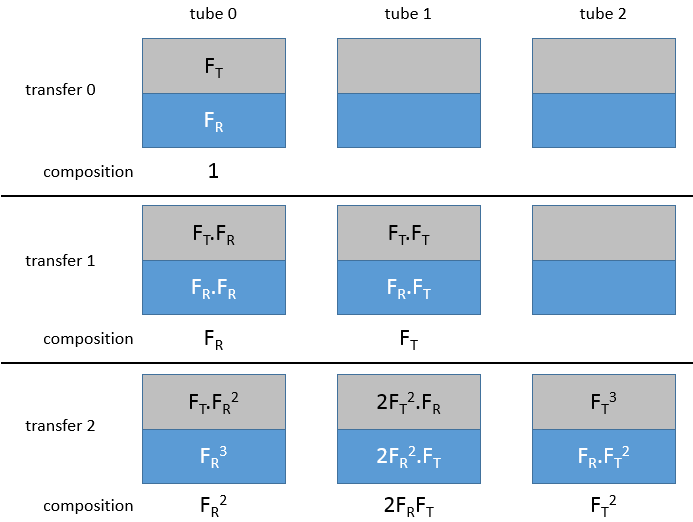
Au début tout le substrat est dans le tube 0, la moitié de celui-ci est dans la phase organique et l’autre moitié dans la phase aqueuse. La phase organique est transférée dans le tube 1. On peut définir une fraction FR restant qui est la fraction de l’échantillon qui reste dans le tube lors d’un transfert et une fraction de transfert FT qui est la fraction qui est transférée dans le tube suivant .. Après le premier transfert il y a FR dans t0 et FT en t1. Le tube 0 est maintenant rempli avec le solvant et les tubes sont mélangés.
Après avoir mélangé le tube 0 la fraction restante FR sera séparée en 2 phases : organique et aqueuse. Dans la phase aqueuse (en considérant que la phase organique est transférée à chaque fois) il restera la fraction restante de la première fraction restante à savoir FR.FR. Dans la phase organique, il y aura la fraction transférée de la fraction restante à savoir FR.FT.
Dans le tube 1 la fraction transférée à partir du tube 0 est séparée entre les deux phases. Dans la phase aqueuse il sera maintenant FR.FT et dans la phase organique FT.FT. Une fois que le mélange est effectué, les phases organiques sont transférées sur le tube suivant. Les phases aqueuses restent dans leur tube. FT2 est transféré de t1 à t2 et FR.FT est transféré de t0 à t1. Après le transfert on trouve respectivement dans les tubes 0, 1 et 2, FR2, 2FR.FT and FT2.
Après trois transferts nous aurions FR3, 3FR2.FT, 3FR.FT2 and 3FR3. Vous avez peut-être remarqué que ces valeurs sont les membres d’un exposant d’une somme :
![]()
n étant le nombre de transfert. Dans un tube r donné la fraction est :
![]()
On peut donc prédire la composition de chaque tube en connaissant le résultat d’une seule extraction. Pour un grand nombre d’extractions nous trouvons l’équation d’une courbe de Gauss.
La largeur du pic est caractérisée par un écart-type :
![]()
Le pic se déplace linéairement avec n répartis comme √n et l’intensité des baisses de pointe en 1 / √n.
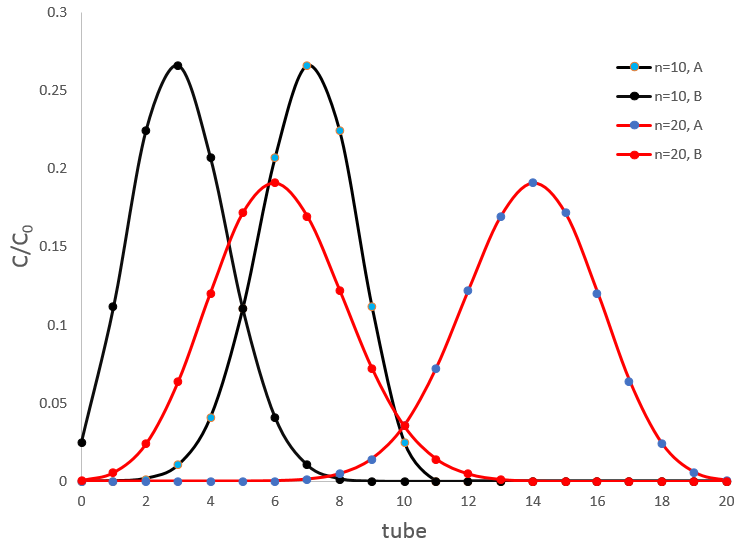
Si deux substrats sont dans l’échantillon l’appareil de Craig va les séparer. Encore une fois nous avons à choisir attentivement les conditions de l’extraction pour obtenir une bonne séparation. La figure ci-dessus montre les concentrations de deux espèces avec DA=1/DB=9/4 après 10 et 20 transferts. Il y a un chevauchement entre les courbes où les espèces ne sont pas bien séparées mais il diminue considérablement avec le nombre d’extractions. Au maximum du pic pour A, après 10 transferts, nous trouvons ~ 27% de la concentration initiale de A mais également 1% de la concentration initiale de B. Après 20 transferts on a 19% de [A]0 et seulement 0,03% de [B]0.
Les avantages de l’appareil de Craig sont que tout est mécanique de sorte que la précision est bonne et que l’on peut obtenir soit le résultat de l’extraction à la fin de l’appareil ou à un tube donné dans l’appareil.
Chapitre 2: thermodynamique – deuxième et troisième principes
Deuxième principe :
Augmentation de l’entropie: L’entropie dans l’univers augmente avec le temps.
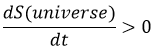
L’entropie est une mesure du désordre d’un système. La seconde loi de la thermodynamique a plusieurs formulations mais la plus commune est qu’un processus est spontané si, sans influence externe (système isolé), il induit une augmentation de l’entropie dans l’univers.
Par exemple, si deux réservoirs sont connectés, l’un étant vide et l’autre plein de gaz, le gaz occupera spontanément tout le système même s’il n’implique aucune variation d’énergie.
Le gaz se dilate car il y a plus de positions à occuper dans un plus grand volume. Une autre définition de l’entropie est qu’un système a tendance à changer à l’état le plus probable.
Imaginez une petite boîte avec seulement quelques positions disponibles. Deux particules sont dans cette boîte et chacune occupe une place. Plusieurs combinaisons sont possibles:
Si nous augmentons le volume de la boîte, la quantité de possibilités augmente exponentiellement. C’est une entropie de position / disposition. La formule de ce type d’entropie pour une particule est :
![]()
Ω étant la quantité de configurations possibles pour cette particule.
Si le volume contenant cette particule passe d’un volume V1 à un volume V2, la variation de l’entropie est
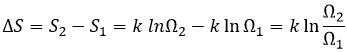
Le nombre de positions pour une particule est directement proportionnel au volume et
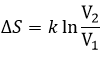
Pour les gaz dilués, le volume des particules est négligeable par rapport au volume du système. En conséquence, nous pouvons ignorer la présence d’autres particules dans la formule et dire que pour les particules NA (1 mole)
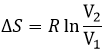
Nous avons vu plus haut la dilatation d’un gaz que l’énergie était une fonction d’état mais que le travail et la chaleur n’étaient pas et donc que la façon dont le processus se déroule a une influence sur le travail et sur la chaleur. Le troisième chemin que nous avons discuté était le processus isotherme pour lequel nous avons trouvé une expression pour le travail
Ce processus est isotherme donc ΔE = q + W = 0.
Nous pouvons donc trouver la variation d’entropie pour ce processus par la chaleur. Si le gaz se dilate, la chaleur pénètre dans le système (c’est un processus endothermique).
La variation de l’entropie est donc équivalente à la quantité de chaleur que le gaz doit absorber pour maintenir sa température constante.
Les états de la matière n’ont pas la même entropie. Un gaz a une valeur d’entropie plus grande qu’un liquide, lui-même un peu plus grande que celle d’un solide. Nous pouvons facilement comprendre cela en utilisant la répartition des particules dans une petite boîte.
Dans un gaz (parfait), les particules n’ont pas d’interactions entre elles. Ils peuvent ainsi occuper n’importe quelle position du volume sans restriction. Dans un liquide, les particules interagissent entre elles et restent groupées mais la forme de la grappe n’est pas figée. Les possibilités sont donc plus limitées que pour un gaz. Dans un solide, les particules sont fixées ensemble dans une forme donnée. Les positions qu’un solide peut prendre sont donc plus limitées que celles qu’un liquide peut prendre et beaucoup plus limitées que pour un gaz.
Des tables d’entropies standard existent et donnent l’entropie des molécules dans un état donné à 298K. Pour résumer, plusieurs cas peuvent être distingués. Un processus se produit si la production d’entropie dans l’univers est positive. Cette production d’entropie est composée de la variation de l’entropie dans le système et dans l’environnement. Une de ces composantes de l’entropie peut être négative si l’autre composante est positive et plus grande en valeur absolue.
Quand une réaction produit un gaz, la variation de l’entropie est positive. Si cette réaction est exothermique, la chaleur est transmise à l’environnement, ce qui signifie que son entropie augmente. Cette réaction est donc spontanée. Si la réaction était endothermique, la variation de l’entropie de l’environnement aurait été négative. La réaction pourrait encore être spontanée si la production de l’entropie de la réaction est plus grande que la diminution de l’entropie dans l’environnement.
Cycle of Carnot
Nicolas Léonard Sadi Carnot a proposé un moyen de convertir l’énergie thermique en travail. C’est un cycle thermodynamique, c’est-à-dire une série de transformations d’état ramenées à l’état initial, composées de deux processus isothermes (pas de variation de température) et de deux processus adiabatiques (pas d’échange thermique avec l’environnement). Tous les processus sont réversibles et le cycle peut donc se faire dans le sens opposé, convertissant le travail en une différence de température.
La configuration est faite d’un réservoir dont le volume peut changer et qui peut être en contact avec deux corps de température T1 et T2, T1 étant plus grand que T2.
Première étape: expansion isothermique du gaz à T1. Le réservoir est en contact avec le corps chaud à T1. La chaleur q1 est échangée entre le corps et le réservoir de gaz qui se dilate sans variation de température. Il y a donc une variation d’entropie ΔS1=q1/T1.
Deuxième étape: expansion adiabatique du gaz. Au cours de cette deuxième étape, il n’y a pas d’échange de chaleur mais le gaz continue à se dilater, à travailler sur le piston et à refroidir de T1 à T2. Le corps à T = T1 n’est plus en contact avec le réservoir. Comme il n’y a pas d’échange de chaleur, il n’y a pas de variation d’entropie.
Troisième étape: compression isothermique du gaz en T2. Le réservoir est maintenant en contact avec le deuxième corps, à T = T2, provoquant un transfert de chaleur q2 tandis qu’une compression est appliquée par le piston, gardant la température constante. Il y a donc une variation d’entropie ΔS2=q2/T2.
Quatrième étape: compression adiabatique du gaz. Le corps à T = T2 n’est plus en contact et il n’y a pas de transfert de chaleur possible. Le gaz continue à diminuer en volume tandis que sa température change de T = T2 à T = T1. A la fin de cette étape, le système est de nouveau dans les conditions (volume, température et pression) de l’état initial.
Le cycle peut être tracé dans le diagramme de phase pV comme indiqué ci-dessous:
L’efficacité R = W/q1 du cycle de Carnot serait de 100% si tout le travail W était transformé en chaleur q. Le travail ici est la différence de chauffage q1-q2 car ΔE = 0 = q + W (système isolé, les corps en T1 et T2 étant dans le système). Par conséquent,
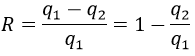
Il est possible de montrer que le rapport q2/q1 est égal au rapport T2/T1.
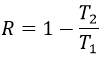
La conséquence est que l’efficacité n’est jamais à 100% sauf à T2 = 0K, que nous ne pouvons pas atteindre sur terre. La perte d’énergie est causée par la production d’entropie.
Energie libre de Gibbs :
Pour qu’une réaction se produise, nous venons de voir que deux facteurs sont importants: l’enthalpie de réaction (si la réaction est exo ou endothermique) et la variation d’entropie. L’énergie libre de Gibbs ΔG regroupe les deux facteurs dans une équation
![]()
Une réaction est spontanée si ΔG <0. Le terme entropique montre clairement qu’une réaction peut être spontanée à une température donnée mais pas si la température est modifiée
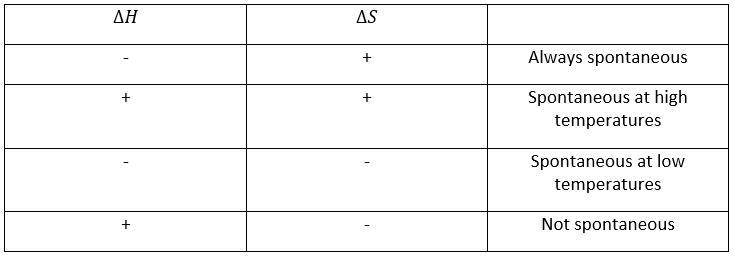
Nous pouvons trouver des tables pour des valeurs standard de ΔH0 et ΔS0 et des tables de ΔG0 pour les réactions de formations de molécules. A d’autres températures mais à 1 atm, la relation est :
![]()
Q est le quotient réactif que nous avons déjà rencontré précédemment :
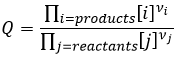
Par exemple, le quotient réactif de cette réaction
![]()
est:
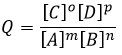
Si AG est négatif, la réaction va vers la droite, si elle est positive, les réactions vont vers la gauche. Si ΔG = 0, nous sommes à l’équilibre. En conséquence, nous pouvons trouver une relation reliant la constante de l’équilibre K avec l’énergie libre de Gibbs:
![]()
Le potentiel chimique :
Le potentiel chimique μi est l’énergie d’une molécule i qui peut être utilisée lors d’une réaction ou d’un processus physique.
Équation de Gibbs-Duhem:
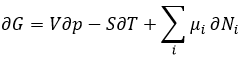
Le dernier terme est donc une variation d’énergie dans le système due à une variation des quantités de particules dans le système, ce dont nous n’avions pas tenu compte auparavant (il sera montré entre parenthèses dans les équations suivantes). La relation ci-dessus vient des définitions de G et de H:
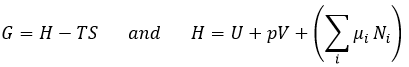
Les dérivées partielles de ces fonctions sont
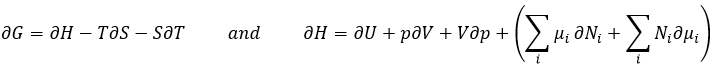
Nous avons vu que l’énergie interne ∂U=∂q+∂W-(∑iNi∂μi) et que ∂S=∂q/T et que ∂W=-p∂V. Par conséquent.
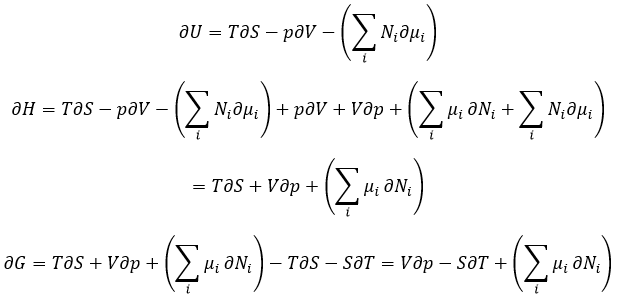
Le potentiel chimique est donc la variation de l’énergie libre de Gibbs en ce qui concerne la variation de concentration d’une espèce spécifique, en gardant les autres paramètres (p, T et les concentrations des autres espèces) constants.
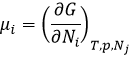
Nous définissons également le volume molaire partiel
c’est utile pour les gaz. De la loi des gaz parfaits,
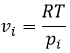
Cela nous conduit à l’expression du potentiel chimique pour un gaz
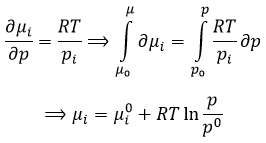
En solution, la formule est très similaire
![]()
Nous savons que
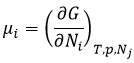
On peut donc trouver l’expression de ΔG en fonction des pressions ou des concentrations de l’espèce:
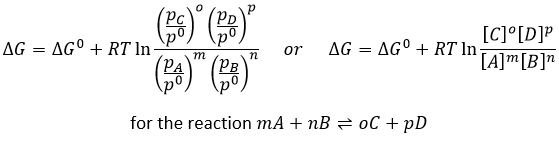
Pourtant, cela n’est vrai que pour les solutions idéales et les gaz. Normalement, nous devons considérer l’activité de chaque molécule
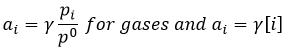
γ est égal à 1 pour les solutions idéales mais peut varier si les concentrations augmentent, ce qui représente l’augmentation des interactions entre les espèces. Cependant, l’activité a des solides est égale à 1.
Troisième principe :
L’entropie d’un cristal parfait au zéro absolu est exactement égale à zéro.
La troisième loi fournit une référence pour l’entropie de tout cristal pur (et donc de n’importe quoi) à n’importe quelle température. Un cristal parfait et pur est un cristal dont l’arrangement est parfaitement organisé: il n’y a pas de défaut, la matrice est régulière et elle est composée d’une seule espèce. Au zéro absolu, c’est-à-dire zéro Kelvin, l’état du système est à l’énergie minimale possible. Cet état est unique du fait de la mécanique quantique et c’est bien le cas: il n’y a qu’une seule façon de placer les atomes dans un cristal parfait. La quantité de configurations possibles est donc 1, conduisant à une entropie égale à
![]()
Si un état de base est dégénéré, c’est-à-dire qu’il y a plusieurs états de même énergie, il y a plusieurs configurations possibles Ω et l’entropie n’est pas égale à 0 mais elle est très proche de celle-ci. Dès qu’il y a un défaut dans le cristal, le nombre de configurations possibles augmente. En conséquence, l’entropie augmente. Cette variation d’entropie est synonyme de variation d’énergie comme le dit la deuxième loi
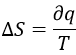
L’augmentation de température dT due à la chaleur ∂Q est déterminée par la capacité calorifique (cp ≈ cV) du cristal
![]()
Conséquences de la troisième loi
Une première conséquence de la troisième loi est que T= 0K ne peut être atteint par un nombre quelconque de processus finis. Cela est dû au fait qu’il n’y a qu’un seul état d’énergie minimale. Si nous essayons de changer de façon contrôlée un paramètre donné pour que l’énergie diminue à chaque fois, il faudrait un nombre infini de répétitions pour atteindre le zéro absolu. C’est un peu similaire au problème de la balle: si à chaque période de temps une balle se déplace de la moitié de la distance nécessaire pour atteindre sa cible, la cible sera approchée mais jamais atteinte. Si plusieurs états étaient autorisés à T= 0K, il serait possible de basculer le paramètre pour obtenir l’un ou l’autre état: une valeur du paramètre s’approche du système d’un état et l’autre approche du système de l’autre état. Avec une seule cible, nous ne pouvons pas passer d’une valeur du paramètre à l’autre et atteindre l’état final. Nous pouvons visualiser ceci comme ceci :
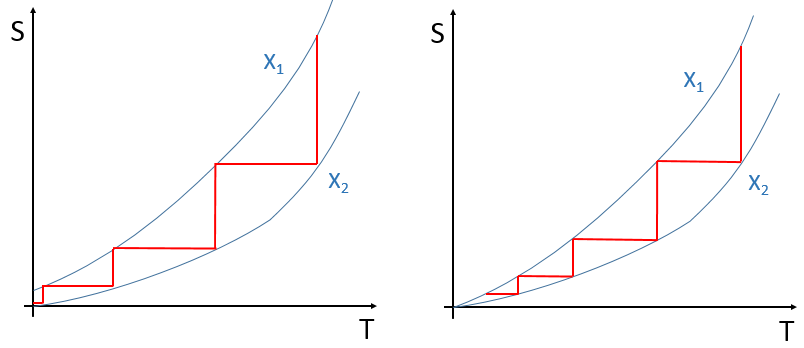
Une deuxième conséquence est que les capacités calorifiques cp et cV tendent vers zéro lorsque T tend vers zéro.
Chapitre 1: Thermodynamique
La thermodynamique est l’étude du transfert de l’énergie et surtout de la chaleur. Il est important de faire la distinction entre la chaleur (q) et la température (T). La température reflète le mouvement des particules et est reliée à leur énergie cinétique mais la température n’est pas l’équivalente de l’énergie. La chaleur est une énergie qui est proportionnelle à la différence de température entre plusieurs compartiments, par convention elle va à partir du compartiment le plus chaud vers celui qui est le plus froid. Les unités de la chaleur sont les unités d’ énergie, c.-à-joules J = Nm
Quand il s’agit de transferts d’énergie nous devons définir le système dans lequel nous travaillons. Le système est la partie de l’univers que nous analysons et nous distinguons à partir de l’environnement qui est le reste de l’univers.
3 types de systèmes peuvent être définis :
– Le système isolé : il n’y a pas d’échange entre le système et l’environnement
– Le système fermé : les échanges d’énergie sont possibles entre le système et l’environnement
– Le système ouvert : les échanges d’énergie et/ou de matières sont possibles entre le système et l’environnement
Un système est exothermique s’il dégage de la chaleur. S’il prend la chaleur, le système est endothermique.
La thermodynamique est régie par trois grands principes.
Premier principe :
Conservation de l’énergie : l’énergie de l’univers est constante.
![]()
Cela signifie que le processus se produisant dans un système isolé ne comporte pas de variation de l’énergie. Si les échanges d’énergie sont autorisés avec l’environnement, par exemple dans des systèmes fermés, la variation interne de l’énergie dans un système est composé par le q de chaleur et le W de travail.
![]()
Si le système permet également les échanges de matière, un terme représentant l’énergie de l’écoulement de la matière dEm est ajoutée.
![]()
La variation de l’énergie est une fonction d’état. Cela signifie que cette fonction ne dépend pas de quelle manière le système a atteint ce point mais seulement sur son état initial et final. W et q ne sont pas des fonctions de l’Etat et dépendent de la façon dont le système évolue au cours du processus.
Le travail a une valeur négative si elle est faite par le système. Par exemple, imaginez un volume dans lequel un gaz est stocké.
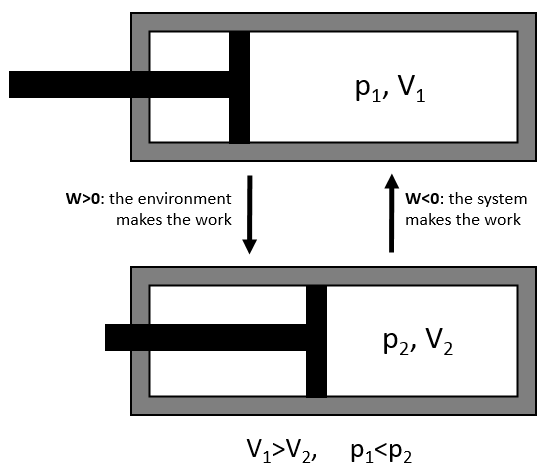
Pour comprimer le gaz l’environnement doit appliquer une force sur le piston. W est donc positif dans ce cas. Si le piston est libre de mouvement, le gaz dans le volume applique une pression sur le piston pour augmenter le volume du système. Dans ce cas W <0.
Le travail est une énergie il est donc essentiellement une force multipliée par la distance. Si nous considérons la dilatation d’un gaz parfait le travail serait :
![]()
Pour passer d’un état (p1, V1) à (p2, V2) plusieurs chemins peuvent être pris.
Ces chemins sont équivalents en terme de variation de l’énergie parce que l’énergie est une fonction d’état : l’état final et les états initiaux sont caractérisés par une énergie qui ne dépend pas de la façon dont ils ont été obtenus. Pourtant le travail et la chaleur sont différents pour chaque trajet.
En effet sur le chemin 1, W = -p1 (V2-V1). Sur le chemin 2 le travail est W = -p2 (V2-V1). Comme p2 <p1, cela signifie que plus de travail est nécessaire sur la voie 1 et qu’il ya une plus grande production de chaleur sur la voie 2. Sur la voie 3 le volume et la pression varient simultanément. Nous utilisons la loi du gaz parfait pour déterminer le travail dans ce cas. Voie 3 est en fait une courbe isotherme où q = 0.
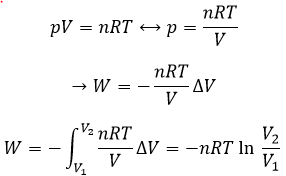
Maintenant parlons un peu de chaleur. Dans les chapitres précédents nous avons discuté à propos de la capacité de la chaleur. C’est l’énergie nécessaire pour augmenter la température du substrat par un kelvin/degré. Nous distinguons la capacité thermique à pression constante Cp et au volume constant CV.

Notez que nous avons utilisé les dérivés partiels ∂ / ∂T au lieu de dérivés d/dt. Un dérivé partiel d’une fonction de plusieurs variables est son dérivé par rapport à une de ces variables, les autres restant constants.
H est l’enthalpie de l’état et ΔH est la variation d’enthalpie du processus (qui peut être une réaction ou une transition de phase par exemple) à une pression constante. ΔH est donc équivalente à qp, l’indice p indiquant que c’est la chaleur à p = cst :
![]()
L’équation de l’enthalpie est :
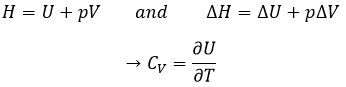
Pour les gaz parfaits PV = nRT donc :
![]()
Étant donné que Cp= ∂H/∂T nous trouvons une relation entre Cp et CV des gaz parfaits :
![]()
Pour les solides et les liquides la variation de volume est négligeable et Cp»CV.
La calorimétrie :
Une enthalpie de réaction AH° est associée à chaque réaction.
![]()
Le ° indique que cette valeur est l’enthalpie trouvée pour une mole de réactif à 1 atm. Expérimentalement l’enthalpie de la réaction peut être mesurée par calorimétrie. Au cours de la calorimétrie une réaction est réalisée dans un système isolé (pas d’échanges avec l’environnement). La variation de la température dans le système est mesurée et à partir de cette mesure nous pouvons déduire la chaleur de réaction. Par exemple, la neutralisation de l’HCl par du NaOH est exothermique (AH°<0) ce qui signifie que cette réaction produit de la chaleur. Il est ainsi possible de mesurer une variation de la température ΔT pendant la réaction. Pour ce faire, nous utilisons un récipient fermé et isothermique dans lequel les réactifs sont initialement séparés. La température dans le système peut être lue sur un thermomètre. La température n’est pas directement prise dans la solution (des réactives). Habituellement la réaction a lieu dans un récipient appelé bombe calorimetrique qui est entouré par l’eau. La configuration entière est dans le recipiant isotherme. La variation de la température est mesurée dans l’eau. Lorsque la réaction a lieu l’élévation de la température est enregistrée en fonction de la quantité utilisée de NaOH. Avec le réglage adapté nous pouvons mesurer qp (p = cst) ou qV (V = cst).
Si la pression est maintenue constante la variation de la température ΔT est proportionnelle à qp.
![]()
Comme nous considérons une phase liquide qp»qV.
CV peut également être déterminée expérimentalement : une quantité de chauffage donnée est appliquée à la solution à travers une résistance électrique :
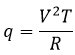
Avec la tension V et la résistance R la variation de température ΔT en raison de ce chauffage dépend de la capacité calorifique du liquide :
![]()
Donc si l’on connaît la tension appliquée et la résistance la mesure de la variation de la température donne la valeur de Cv.
![]()
Loi de Hess :
L’énergie et l’enthalpie sont des fonctions de l’état, à savoir que leur valeur ne change pas en fonction de la voie/processus et ne sont qu’une fonction de l’état du système. Cependant la loi de Hess dit que la valeur absolue de l’énergie des choses ne peut pas être déterminée seules les variations de l’énergie peuvent être déterminées. Cela signifie que l’on peut déterminer la valeur de l’enthalpie par rapport à un procédé donné et donc que l’enthalpie de formation d’une molécule est déterminée par rapport aux autres réactions.
Par convention, l’enthalpie de formation de corps simples dans leur état standard à 25°C et 1 atm est égale à 0 kJ/mole. Dans la nature certains éléments peuvent être trouvés dans plusieurs Etats mais le corps dans son état normal à 25°C et 1 atm seul a une ΔfH° = 0 kJ / mole. Par exemple
By convention, the enthalpy of formation of simple bodies in their standard state at 25°C and 1 atm equals 0kJ/mol. In the nature, some elements can be found in several states but the body in its normal state at 25°C and 1 atm alone has a ΔfH°=0kJ/mol. For instance,
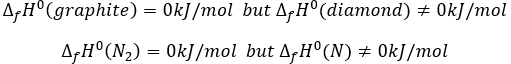
Pour les molécules qui ne sont pas dans leur forme standard l’enthalpie de formation est l’enthalpie de la réaction produisant la molécule à partir des éléments dans leur état standard qui le composent.
![]()
L’enthalpie de cette réaction est donc l’enthalpie de formation du dioxyde de carbone CO2 : il est la réaction qui génère du dioxyde de carbone à partir des éléments dans leur état standard qui composent CO2.
![]()
Cette réaction est l’inverse de la formation du méthane CH4. L’enthalpie de réaction est simplement l’enthalpie de formation avec un signe(-) ΔrH°=-ΔfH°.
![]()
L’enthalpie de cette réaction est deux fois l’enthalpie de formation de l’eau parce que nous formons deux moles d’eau avec cette réaction. L’enthalpie de réaction est donnée en kJ par mole de produit.
Si nous combinons ces trois réactions nous obtenons le cycle de Hess. L’enthalpie de cette équation finale est la somme des enthalpies de formation des trois sous-réactions :
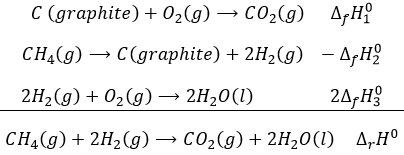
On peut résumer les enthalpies des réactions secondaires pour obtenir l’enthalpie de la réaction de formation de CO2 et H2O de CH4 et O2.
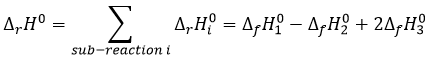
D’une manière générale, l’enthalpie de réaction est la somme des enthalpies de formation des produits moins les enthalpies de formation des réactifs chacun multiplié par ses coefficients stoechiométriques.
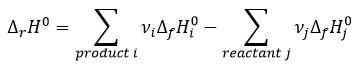
Nous voyons clairement ici que ces deux expressions sont identiques :
![]()
Ne pas oublier que l’enthalpie est une fonction de l’état, mais dépend de la température et que la réaction peut être favorisée par une variation de la température.
![]()
À une pression constante l’enthalpie peut donc varier en cas de variation de température
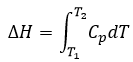
Un changement d’état va également augmenter l’enthalpie de la réaction.
Si nous voulons changer la glace en vapeur, à savoir sublimer la glace, à une pression constante = 1 atm,
![]()
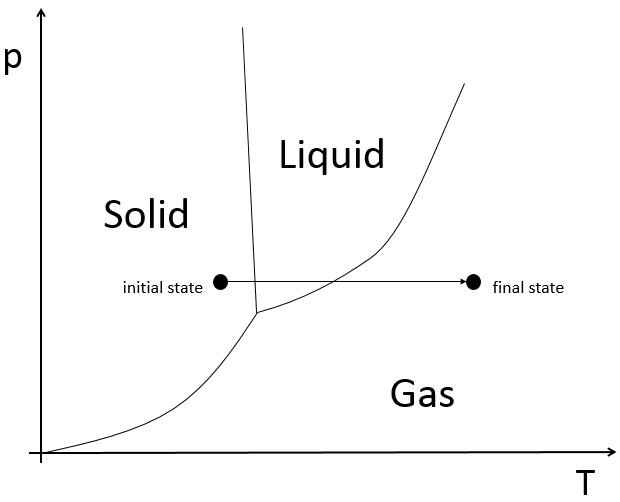
L’enthalpie sera
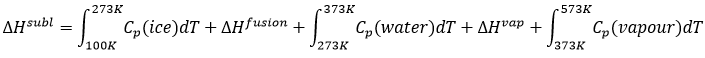
Le point triple est un point particulier où la glace, la vapeur et l’eau coexistent et où :
![]()
De ce point nous pouvons trouver l’enthalpie d’une transition de phase des deux autres.
L’énergie de liaisons :
Nous avons vu que nous pouvons trouver l’enthalpie deS réactions des enthalpies de formations de chaque participant de la réaction. Il devrait donc y avoir des réactions qui nous permettent de déterminer l’énergie de liaisons chimiques. Par exemple la réaction :
![]()
est fondamentalement la scission de 4 liaisons C-H. Rien d’autre n’a été fait au cours de cette réaction. On peut déterminer l’enthalpie de la réaction ΔrH0 de l’enthalpie de formation de CH4, C et H. On a vu plus haut l’inverse de la réaction de formation du méthane
![]()
De cette réaction nous avons encore besoin de dissocier les molécules de H2 et de sublimer le graphite dans un gaz.
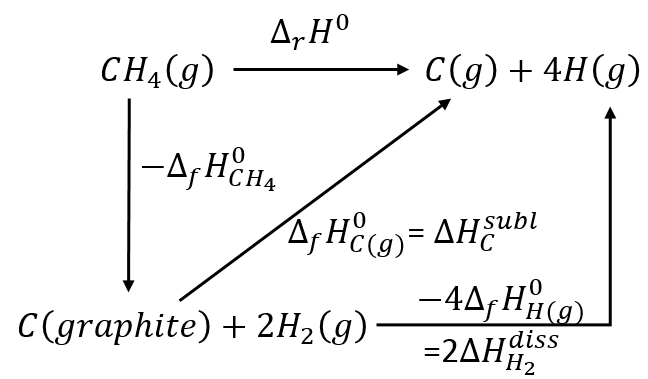
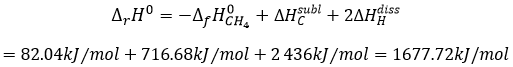
Comme il ya quatre liaisons CH qui ont été brisées chaque obligation a une énergie de 419kJ / mol.
Carburants :
La thermochimie est un vaste domaine de la chimie qui trouve des applications dans les carburants par exemple. Nous pouvons déterminer l’énergie des liaisons lors de la combustion des molécules composant le carburant exactement de la même manière que nous l’avons fait pour la liaison C-H. L’hydrazine N2H4 est une molécule d’azote utilisée comme carburant pour les hélices de l’espace.
![]()
La plupart des composés azotés ont une enthalpie positive de la formation ce qui signifie que nous avons besoin de dépenser de l’énergie pour les générer. Cela signifie aussi qu’ils vont se casser facilement pour former N2 et H2O générant beaucoup d’énergie. Une exception est NH3. Quel est l’énergie de la réaction? O2 et N2 sont dans leur forme standard ce qui signifie que leur ΔfH0 = 0kJ/mol. L’enthalpie de réaction est ainsi
![]()
Cette réaction produit de la chaleur parce que l’énergie des liaisons formées est supérieure à l’énergie des liaisons cassées. Nous avons cassé 4 liaisons NH pour former 4 OH et la liaison NN devenue une liaison triple N≡N. Les liaisons OH et N≡N représentent respectivement 460kJ/mole et 941kJ/mole.
En raison de la quantité d’énergie qui a été générée par la réaction, il y a un changement de la phase et l’eau devient gaze. La propulsion de la navette spatiale est le résultat de l’énergie,qui provient de l’expansion du gaz : à partir d’une mole d’eau on fait 3 moles de gaz. Le gaz s’élargit très rapidement en raison de la chaleur (pV = nRT) générant l’explosion dans l’hélice.
Cette réaction produit de la chaleur parce que l’énergie des liaisons formées est supérieure à l’énergie des liaisons cassées. Nous avons cassé 4 liaisons NH pour former 4 OH et la liaison NN est devenue une liaison triple N≡N. Les liaisons OH et N≡N représentent respectivement 460kJ/mole et 941kJ/mole.