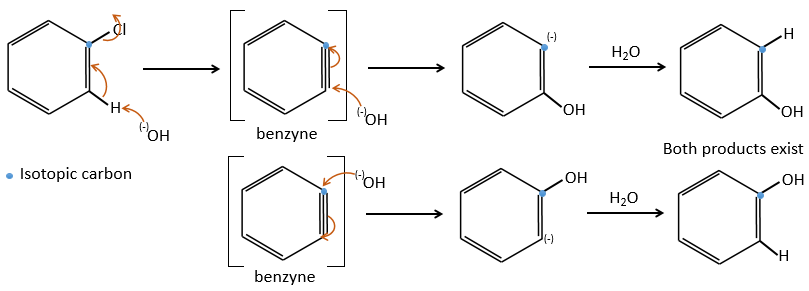
Chapitre 10 : chimie physique moléculaire – opérateurs
Les opérateurs peuvent être appliqués aux fonctions d’onde et respecter l’équation de Schrödinger. L’inversion de l’opérateur Î est un opérateur tel que, si une symétrie centrale peut être trouvée,
![]()
Par exemple, l’orbitale s a un centre de symétrie. Nous disons que cet état est pair. Si nous appliquons l’opérateur d’inversion à cette orbitale, le signe de l’orbitale est toujours inchangé du côté opposé de l’atome (c’est-à-dire obtenu par symétrie centrale).
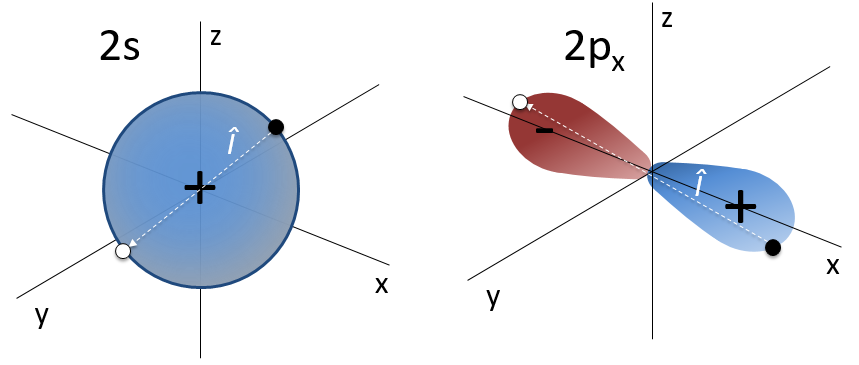
L’orbitale p est impaire parce que si nous regardons un point dans la partie positive de l’orbitale et appliquons la symétrie, nous serons dans le lobe négatif de l’orbitale. Les orbitales d sont paires. En fait, nous avons
![]()
L’opérateur Î peut également être appliqué aux électrons pour obtenir le spin opposé (en haut ou en bas).
Le moment angulaire L est le produit croisé de deux vecteurs r et p. Le produit croisé ou produit vectoriel est une opération binaire sur deux vecteurs et est désigné par le symbole ×. Étant donné deux vecteurs linéairement indépendants a et b, le produit croisé, a × b, est un vecteur c perpendiculaire aux deux et donc perpendiculaire au plan qui les contient (deux vecteurs linéaires partagent toujours un plan).
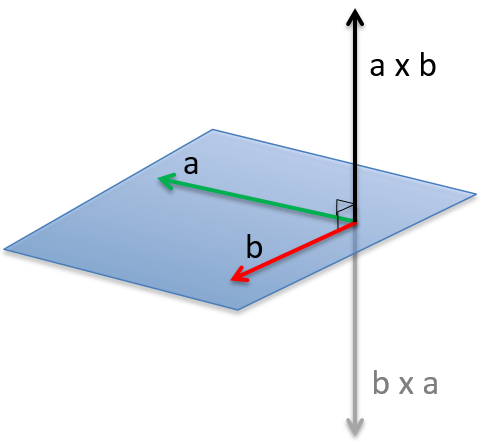
L’intensité du nouveau vecteur est a × b = |a||b| sin n (avec n le vecteur d’unité pointant dans la bonne direction) et la direction du produit croisé est donnée par la règle de la main droite: l’index se tient dans la direction du premier vecteur et le majeur dans la direction du second vecteur. Si vous placez votre pouce perpendiculairement aux deux autres doigts (pour montrer votre approbation), il indique la direction du produit croisé. Il peut être nécessaire de faire tourner votre main étrangement pour obtenir les bonnes directions, par exemple pour obtenir le b x un produit d’en haut.
En mécanique quantique, p est un opérateur
![]()
Nous avons donc
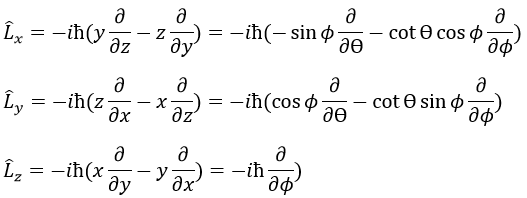
Notez que Lz ne dépend pas de Θ et il y a donc une symétrie autour de l’axe Z pour cet opérateur. Si on applique l’opérateur Lz à la fonction d’onde angulaire, on obtient la fonction d’onde multipliée par ћm:
![]()
Cela signifie que la fonction d’onde angulaire est la fonction propre de l’opérateur Lz. C’est aussi le cas avec l’opérateur L2.
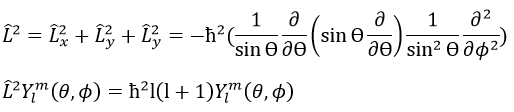
Nous pouvons donc déterminer les valeurs propres de L2 simultanéments(=ћ2l(l+1)), charactéristique du moment angulaire L, et de Lz (=ћm), la projection de L sur l’axe z, pour valeurs fixes des nombres quantique.
La valeur propre d’un opérateur est l’intégrale de la fonction d’onde multiplié par l’opérateur agissant sur la fonction d’onde :
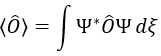
Et la longueur du vecteur L et de ses projections sur les axes :
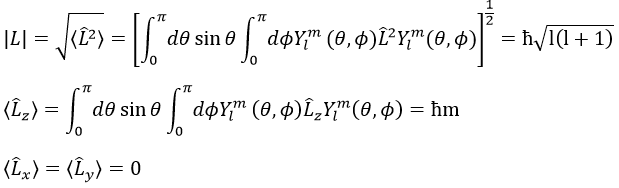
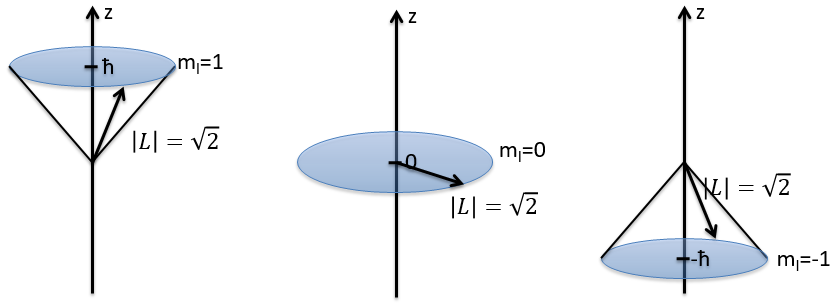
Par exemple, pour l = 1, on peut dessiner L et sa projection en fonction de m. La longueur de L ne dépend pas de m.
L2 et Lz commute: deux opérateurs qui ont la même fonction d’onde. La commutativité signifie que :
![]()
C’est en effet le cas : [L2,Lz]=0 et ce n’est pas le cas entre Lx, Ly et Lz:
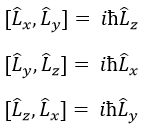
Notez que Lx et Ly aussi commute avec L2, comme Lz fait, mais leur valeur propre <Lx> et <Ly> est égale à zéro.
Le même type de propriétés est vrai pour le spin des électrons avec les opérateurs S2 et Sx, Sy, Sz.
Pour décrire l’ensemble du système, nous avons des séries d’équationscontenant les 5 nombres quantiques n, l, ml, s et ms. Ils forment un CSCO(complete set of commuting observables) .
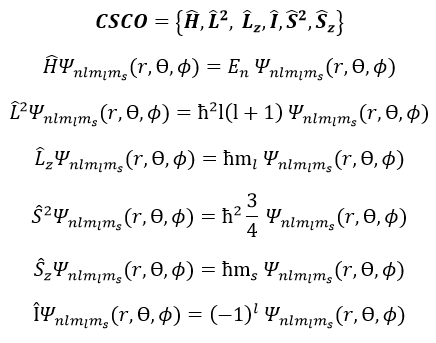
Représentation matricielle :
Dans une base orthonormée de fonctions {Ψ1, Ψ2,…ΨN}, l’opérateur  a une représentation matriciellethe (Â) qui est une matrice composée des opérateurs Âij come.
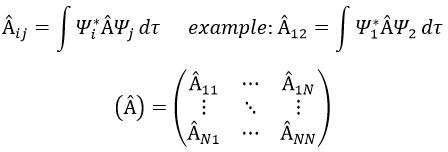
si Ψ1=Ψ2, le résultat aurait été la bonne valeur de Ψ1 si Ψ1 est une fonction propre de Â. En générale on utilise la notation bra-ket de Dirac:
![]()
La partie avant l’opérateur est appelée bra et la partie après l’opérateur est le ket.
si Ψ1 n’est pas une fonction propre de  (ÂΨ≠aΨ), alors nous pouvons trouver une combinaison linéaire (combili) de fonctions d’onde telles que :
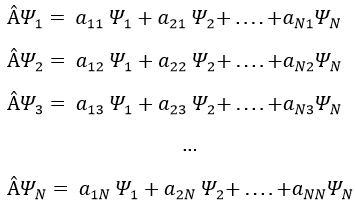
ou écrit en tant que matrices
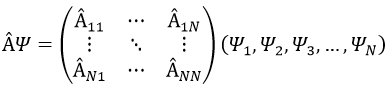
Prenons un exemple avec les opérateurs Lz and L2 et les fonctions angulaires Yml que nous avons utilisées auparavant pour l=1. La base complète des fonctions est Y1m={Y11, Y10, Y1-1}. Les équations sont:
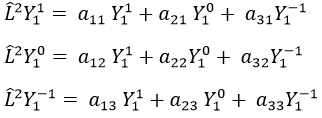
Les valeurs de aij peuvent facilement être trouvées
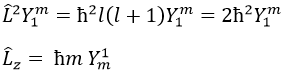
L’équation pour Y11 ne dépend pas de Y1-1 ou Y10. En conséquence, aij=0 for i≠j et aii=2ћ2.
![]()
La même chose vaut pour Y01 et Y11
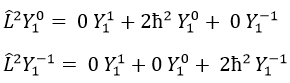
Ensuite, nous avons que la représentation de la matrice (L2) de l’opérateur L2
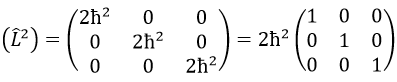
Cette matrice est diagonale, c’est-à-dire que les composants de la matrice qui ne sont pas sur la diagonale sont tous égaux à zéro. (Lz) est aussi diagonale.
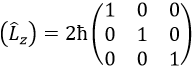
La règle est que si les opérateurs sont des opérateurs de fonctions qui sont des fonctions propres à cet opérateur, alors la matrice est diagonale. Tous les opérateurs de la CSCO ont une représentation matricielle diagonale sur la base de leurs fonctions propres communes. Notez que cela signifie que Lx et Ly ne font pas partie du CSCO: ils ne commutent pas avec Lz et leur représentation matricielle est
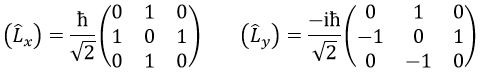
C’est une conséquence directe du principe d’incertitude de Heisenberg: nous ne pouvons pas connaître la position exacte et la vitesse d’une particule en même temps. Par conséquent , L2 ne peut pas commuter avec tous les Lx, Ly et Lz.
On peut définir les opérateurs d’escalade L+ et de descent L– de Lx et Ly.
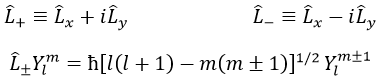
Pour notre exemple avec l = 1, ils sont :
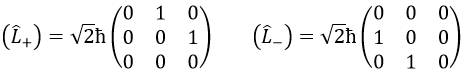
Leur rôle est de passer d’une orbite à l’autre.
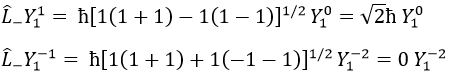
Comme le montre la dernière ligne, il est impossible de sortir du système. Il n’y a pas d’orbitale Y1-2.
Chapitre 9 : Chimie physique moléculaire – l’hydrogène
Pour commencer doucement, nous décrirons la molécule non chargée la plus simple: l’hydrogène. Il est composé d’un proton avec une charge positive e et un électron de charge opposée -e qui tourne autour du proton à une distance r.
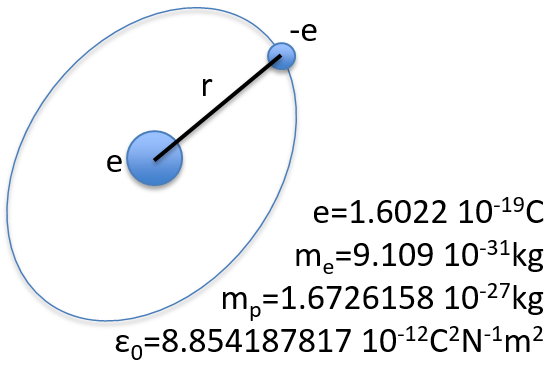
En mécanique quantique, le système est décrit par l’équation de Schrödinger
![]()
Ψ est une fonction d’onde et Ĥ est l’hamiltonien, composé d’un terme cinétique et d’un terme potentiel.
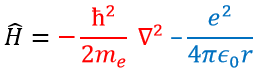
Le terme potentiel est le potentiel d’attraction entre deux charges opposées dans le vide (ϵ0 est la permittivité du vide).
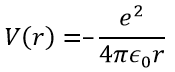
Le terme cinétique décrit le mouvement de l’électron en orbite autour du noyau, où ћ=h/2π et le gradient ∇ est
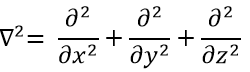
en coordonnées cartésiennes. Il est intéressant de changer pour les coordonnées sphériques. L’expression semble plus complexe mais permet de séparer certaines expressions.
Alors
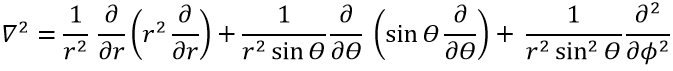
Cette expression semble plus complexe que la précédente, mais il y aura un gros avantage. L’équation de Schrödinger devient
![]()
Cette équation, développée, donne
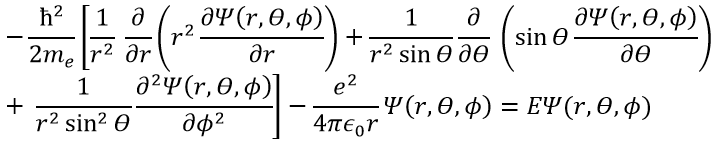
En conséquence, la fonction d’onde peut maintenant être séparée en une équation radiale et une équation angulaire.
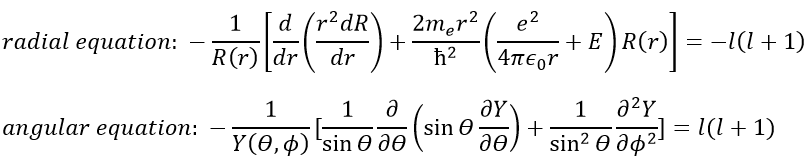
La solution des équations est directement liée au nombre quantique l. Pour rappel, les nombres quantiques n, l et m donnent la configuration électronique des atomes avec l entre zéro et n (O ≤ l ≤ n) et m entre -l et l (-l ≤ m ≤ l). Par exemple si n = 2, l peut avoir une valeur de 1 ou 0, donnant 3 valeurs possibles pour m: -1, 0 (deux fois), 1. Fermi nous dit que nous pouvons mettre 2 électrons de spin opposé sur chaque orbitale. Pour les atomes, les orbitales sont notées s, p, d, f, g, … pour l = 0, 1, 2, 3, 4, … précédées du nombre quantique n.

Nous allons d’abord essayer de résoudre l’équation radiale. Pour ce faire, nous multiplions tout par ћ2/2mer2 et changeons la variable de R(r) to P(r)=rR(r).
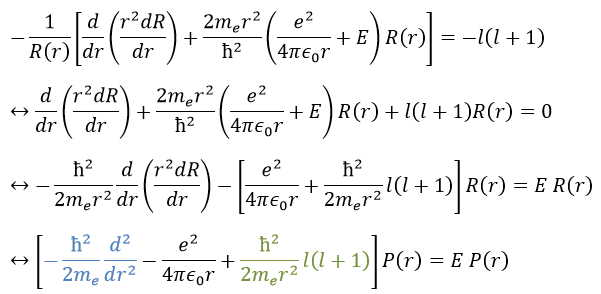
Il y a trois termes dans l’équation: l’énergie cinétique radiale, le potentiel de Coulomb et le terme centrifuge. L’équation est similaire à l’hamiltonien initial, mais il existe un terme de centrifugation supplémentaire. Le terme potentiel est maintenant composé d’un terme de Coulomb (potentiel d’attraction) et du terme centrifuge.
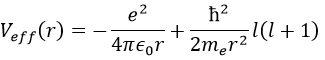
Si nous considérons le potentiel effectif, nous voyons que le terme centrifuge repousse les électrons du noyau. Les orbitales s (l = 0) n’ont pas de terme centrifuge et leurs électrons sont donc proches du noyau. Les autres orbitales impliquent un terme centrifuge positif éloignant les électrons du noyau.
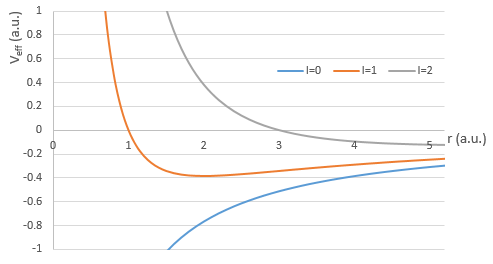
L’énergie En des orbitales dépend de n et pas de l ou m.
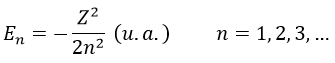
Elle est proportionnelle à 1/n2. Les orbitales sont donc de plus en plus proches les unes des autres quand n augmente. L’énergie est négative et tend vers zéro pour n → ∞, c’est-à-dire l’ionisation. L’énergie est également proportionnelle au carré du nombre atomique. L’énergie est donc différente pour H ou He+, ou n’importe quel atome avec un seul électron malgré la structure similaire.
La solution pour la fonction d’onde est :
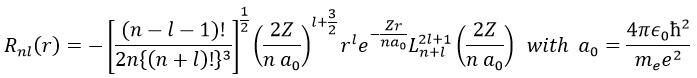
La fonction d’onde Rnl(r) dépendra donc des nombres quantiques n, l, du nombre atomique et du rayon. Dans le cas des orbitales s, la fonction d’onde n’est pas égale à zéro à r = 0. Pourtant, la densité de probabilité est r2.Rnl(r)2 et il n’y a donc pas d’électron dans le noyau.
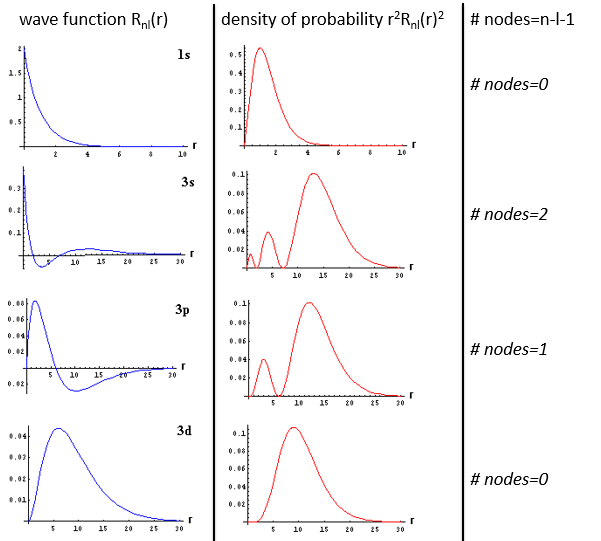
Comme la densité de probabilité est le carré de quelque chose, les zones avec une valeur négative de Rnl(r) peuvent posséder des électrons. Les points où la densité de probabilité est égale à zéro sont appelés nœuds et leur nombre est égal à n-l-1. Notez qu’il n’y a donc pas de rayon fixe pour un électron mais pour sa plus grande probabilité de présence et que même si l’énergie des orbitales est égale, les électrons ne sont pas à la même distance du noyau. Pour les atomes mono-électroniques, les orbitales 3s et 3p sont donc à la même énergie. Ils sont dégénérés. C’est le cas pour toutes les orbitales de même valeur de n.
Maintenant, regardons la fonction d’onde angulaire Y.
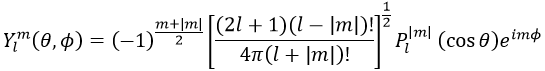
où Pl∣m∣(cos) sont les fonctions associées de Legendre. La solution ici ne dépend pas de n mais de l et m. Quelques solutions sont données ci-dessous:
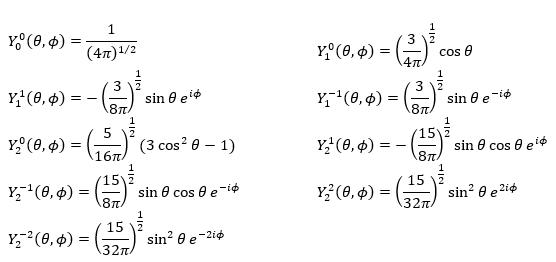
La première fonction d’onde angulaire Y00 donne une sphère car elle n’a aucune dépendance sur les angles et ϕ . Les autres fonctions ont des formes différentes qui dépendent des angles.
Pour obtenir des orbitales atomiques, il suffit de combiner les fonctions d’onde angulaire et radiale. Les orbitales sont sphériques. Il y a 3 orbitales p car il y a 3 valeurs possibles pour m: -1, 0 ou 1. Les orbitales sont identiques mais pointent dans des directions différentes. En fait c’est un peu plus compliqué: les fonctions d’onde angulaire pour m = -1 et 1 ont un terme imaginaire dont on se débarrasse par un changement de coordonnées, revenant aux cartésiennes. Nous pouvons faire ce changement avec une combinaison des orbitales. Nous pouvons dessiner les orbitales avec la technique suivante:
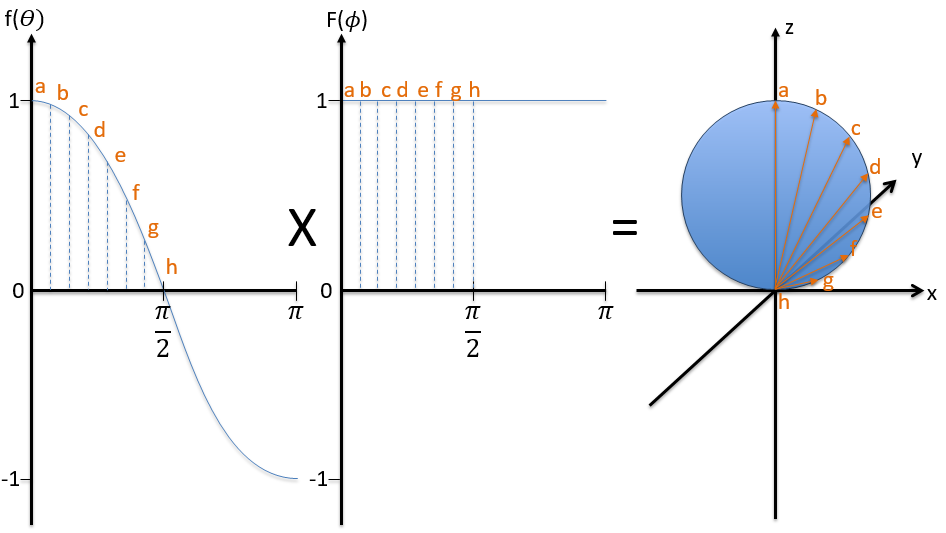
Nous prenons d’abord les fonctions qui dépendent d’un angle et dessinons leur valeur en fonction de l’angle. Pour l’orbital pz les fonctions sont f()=cos et F(ϕ)=1. Quand F = 1 pour n’importe quel angle, f va de 1 à -1. L’orbitale est donnée par la multiplication des deux fonctions. F = constant signifie que l’orbitale est identique pour toute valeur de ϕ. L’axe z est donc un axe de symétrie et l’angle dans la direction z. Quand = 0 (point a), nous sommes sur l’axe z et l’intensité est 1. Quand> 0, nous ne sommes pas sur l’axe. L’intensité est inférieure à un, donc nous dessinons un point à une plus petite distance du centre que le point précédent. L’intensité continue de diminuer pour atteindre zéro à=π/2 (point g), l’intersection avec le plan x = 0, y = 0. En fait, nous avons dessiné une sphère au-dessus du plan x = 0, y = 0. La même chose est faite de l’autre côté du plan (π/2 <<π). Cette partie de l’orbitale a un signe négatif car la fonction est négative (cos <0). Nous pouvons répéter cette méthode pour les autres directions.
Chapitre 8 : spectres de masse – exercices
The solutions immediately follow the problems. You can use this website to find fragments corresponding to a given m/z ratio: MS fragments. The methodology to obtain the answer is given.
Problem 1
The two following spectra come from two isomers with the formula C10H14. Determine the structure of each of them.
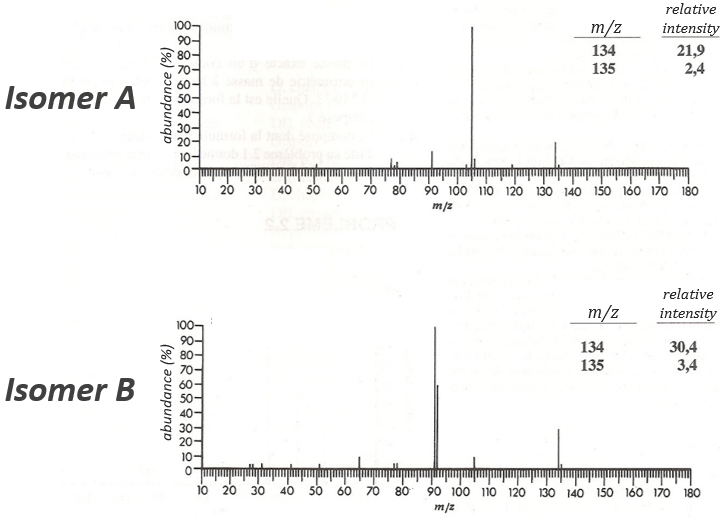
Answer
The isomer A is the sec-butylbenzene (or 2-phenyl-butane) and the isomer B is the n-butylbenzene (or phenylbutane).
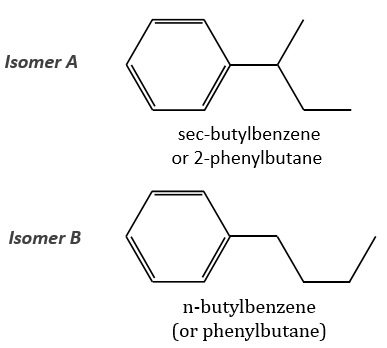
Explanation
The composition of our unknown species is given in the wording: C10H14. The first thing to do is thus to determine the degree of unsaturation of the species.
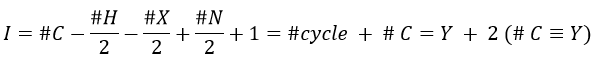
In our case I=4, indicating a phenyl group somewhere in the molecule. It is confirmed by the presence of a peak at m/z=77 (benzene, often with a cluster at 78 and 79) and at 91 (tropylium) on both spectra. The peak of the tropylium at m/z=91 is way more intense for the isomer B than for the isomer A, indicating that the formation of ϕ-CH2+ is somehow less stable in the case of the isomer A. Indeed, the tropylium ion usually shows a very intense peak as we can see on the spectrum of the isomer A.
In our case I=4, indicating a phenyl group somewhere in the molecule. It is confirmed by the presence of a peak at m/z=77 (benzene, often with a cluster at 78 and 79) and at 91 (tropylium) on both spectra. The peak of the tropylium at m/z=91 is way more intense for the isomer B than for the isomer A, indicating that the formation of ϕ-CH2+ is somehow less stable in the case of the isomer A. Indeed, the tropylium ion usually shows a very intense peak as we can see on the spectrum of the isomer A. It is the main difference on which we will base our analysis.
Removing the phenyl from the composition, we find the composition of a butyl group (C4H9). The question is how those 4 carbons are placed around the phenyl but we have already limited by a lot the number of possibilities. We can assume that the 4 carbons make one chain bound to the phenyl and not 2 or more for the isomer B. Let’s focus on this isomer for now. There are 4 possibilities to bind a butane and a phenyl together:
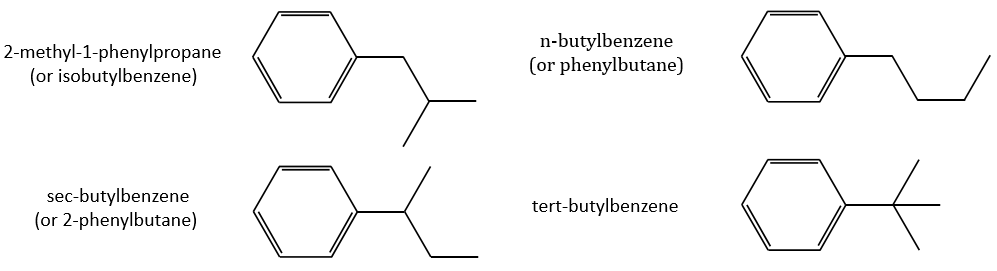
From the intensity of the peak at m/z=91, we can reject the possibilities of the bottom. The two last possibilities, the n- and iso-butyl~ have very similar spectra and both can explain the peak at m/z=92. This peak comes from a rearrangement of Mc Lafferty transferring one proton from the carbon in γ of the phenyl.
We will favour the n-butyl~ because there is a peak of good intensity at m/z=105, i.e. +14 to the tropylium that is hard to explain (and almost inexistent) in the case of the iso-butyl~.
The isomer A can be the sec-butylbenzene that was on our list of 4 structures matching the binding of a butane with a benzene. The obtaining of a tropylium is hard in this case because there are multiple liaisons in α of the phenyl. Moreover, the fragmentation favours the removal of the most substituted fragments from the chain if there are several substituents. The phenyl can be removed (but aromatics are usually not removed from fragments) but also the ethyl chain, what leads to a peak at m/z=105, i.e. the main peak of our isomer A. We can also note the presence of a small (yet existing) peak at M-15. It means that there is a methyl substituent that can be removed. It is less probable than the removal of the ethyl but it can occur. It is thus a good call.
A second possibility is that there are two chains bound to the phenyl, one methyl and one propane. The methyl substituent would the displacement of the tropylium peak by Δm/z=+14 but that’s not how it works. The ion formed would not be as stable as the tropylium and the peak would not be that intense.
The isomers A and B are thus the sec-butylbenzene (or 2-phenylbutane) and the n-butylbenzene (or phenylbutane).
Problem 2
Determine the structure of the compound the spectrum of which is below.
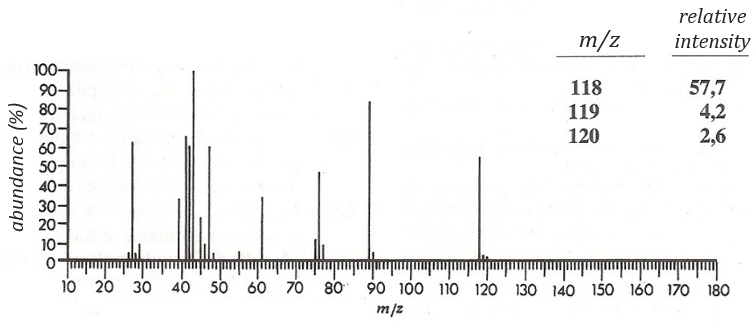
Answer
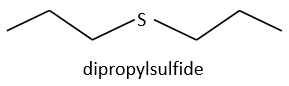
Dipropylsulfide (iupac: 1-(Propylsulfanyl)propane)
Explanation
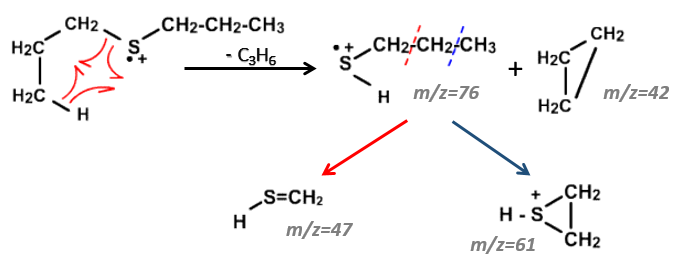
The first thing we (always) do is to look at the parent peak. The mass (M=118) is even, what means that there is an even number of nitrogen in the molecule. More interesting, there is a peak at M+2. Its intensity is about 4.5%, meaning that there should be a sulphur atom in the molecule. If we remove the mass of an atom of sulphur (M=32) from the mass of the compound, we obtain 74, i.e. the mass of C6H14. The M+1 peak is approximatively 7% of the parent peak, in good agreement with the 6 carbons of our guess (1.1% by carbon à 6.6%). Just from the parent peak and the M+1 and M+2 peaks, we have found the composition of the compound. Now, there are a lot of possibilities to place a sulphur in an alkyl chain. So far we just now that there is no cycle in the molecule (otherwise we would have 2H less).
We will thus analyse the fragmentation peaks. The base peak is at m/z=43 and is surrounded by a cluster of intense peaks. The second most intense peak is at m/z=89, i.e. M-29. It corresponds to the loss of a -C2H5 fragment. The liaison between the sulphur and a carbon is usually not cleaved so we can assume that at one side of the sulphur, there is at least a chain of 3 carbons. This fragment is particularly well stabilised because of the possible cyclisation of the ion.
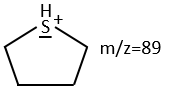
The presence of this intense peak limits a bit our range of possibilities from some substituted chains. In all honesty, it is difficult to explain the exclusion of all the species and why the dipropylsulfide is the correct answer but we will do our best.
The first compound we want to try out is the primary sulphide.
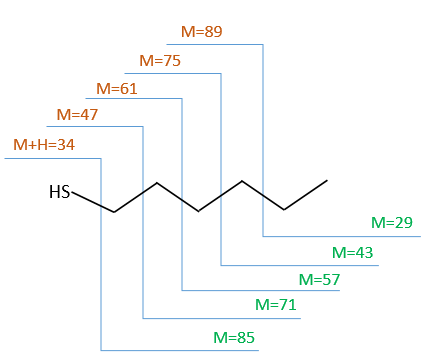
Several things may let us think it is the correct call:
- the cleavage in αβ gives an intense peak at m/z=47.
- the cleavage in βγ gives a peak at m/z=61 the intensity of which is usually the half of the intensity of the peak at m/z=47.
- the cleavage in γδ gives a small peak at m/z=75.
Normally the cleavage in δε gives a peak a bit more intense than at m/z=75 because of the cyclisation of the ion but it is very intense in our spectrum. Moreover, primary sulphide give an intense peak at m/z=34, corresponding to SH2, which is not present at all on the spectrum.
The secondary and tertiary sulphides can be dismissed because of the very strong intensity of the m/z=89 peak. In general, they eliminate the biggest fragment and a very intense peak can thus be observed at M-15, M-29, M-43, etc. Note that we have a very intense peak at M-29. However a peak is usually observed at M-33 (loss of -SH) and there is no trace of it on our spectrum.
We can thus turn our mind towards the thioalkyls. A cleavage gives the very intense peak at M-29. If we assume that the intensity of the peak is similar than for the primary sulphide, we can guess that the m/z=89 peak corresponds to the cleavage of the αβ liaison. There is thus a side of the sulphur with a length of 3 or more carbons. For those thioalkyls, we find a peak at m/z=61which corresponds to the following fragmentation:
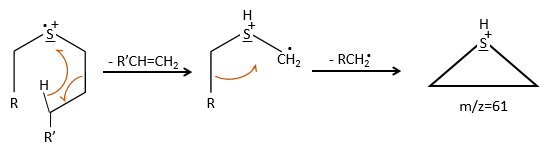
On the spectrum of thioalkyls, we can find peaks corresponding to the cleavage of the liaison S-C, for both the fragment containing the sulphur RS+ but also for the alkyl chain R’+. If the chain is long, the peaks of the alkyl chain can dominate. The base peak of the spectrum is at m/z=43, i.e. C3H7+. This very large intensity can be explained by the fact that the cleavage at both sides of the sulphur in the dipropylsulphide gives this ion. The RS+ peak should be characteristic at m/z=75 but the peak is only of average size. However, there is an important peak at m/z=76. It comes from a particular cleavage leading to the ion RSH+.
Chapitre 7 : Proton RMN: principes
Une section complète peut être (et sera) dédiée à la mise en place détaillée de la résonance magnétique nucléaire (RMN) et aux analyses que nous pouvons effectuer avec cet outil. Dans ce chapitre, nous n’avons pas besoin de beaucoup d’explications sur la théorie de la RMN car c’est une section orientée technique. Nous donnerons cependant une introduction sur la RMN appliquée au proton mais gardons à l’esprit que beaucoup plus peut être fait.
Principe de la RMN
De même que pour la spectroscopie, lorsqu’un champ magnétique est appliqué sur un échantillon, une partie des rayonnements électromagnétiques peuvent être absorbés, dont les fréquences dépendent des propriétés de l’échantillon. Un spectre RMN donne des pics d’intensités diverses en fonction de la fréquence d’absorption. Les atomes ont des réactions différentes résultant de l’application du champ magnétique et absorbent dans des régions séparées de fréquences ou n’absorbent pas du tout. Mais plus important encore, et c’est ce qui nous intéresse dans cette section, la fréquence d’absorption d’un atome dépend de son environnement direct. Par exemple, nous pouvons détecter la différence d’absorption pour les atomes d’hydrogène situés à l’extrémité de la chaîne et à l’intérieur de la chaine dans la molécule suivante CH3-CH2-CH3 (δ≈0.91, 1.34, 0.91).
Premièrement, revenons à l’absorption de différents atomes. Tout noyau porte une charge électrique mais dans certains d’entre eux cette charge tourne dans une direction donnée autour de l’axe nucléaire. Ce mouvement induit un dipôle magnétique qui peut être décrit en terme de nombre de spin nucléaire I. Je peux avoir n’importe quelle valeur positive à partir de 0 et par étape de ½, soit 0, ½, 1, 3/2, 2, … Un isotope a une valeur de spin qui dépend de la masse atomique et du nombre d’isotopes. Nous pouvons reprendre de cette façon:
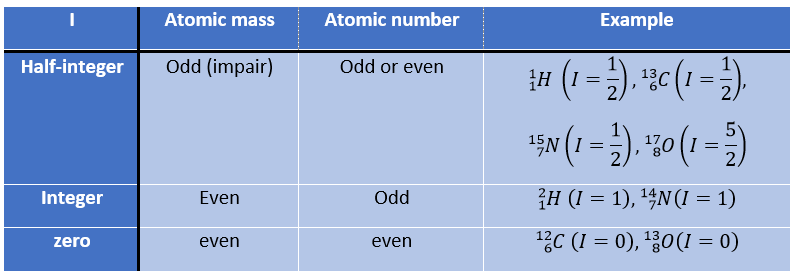
Le nombre de spin détermine le nombre d’orientations qu’un noyau peut prendre dans un champ magnétique externe uniforme, suivant la formule n = 2I + 1. Un atome avec I = 0, tel que le carbone 12, ne peut donc prendre qu’une orientation et est « inactif » en RMN. Les atomes avec I≥1 ont une répartition non sphérique de la charge décrite par un moment quadripolaire qui affecte les noyaux voisins.
Les atomes avec I = 1/2, tels que 1H et 13C peuvent prendre 2 orientations avec des énergies différentes correspondant au spin +1/2 et -1/2. Le niveau d’énergie inférieure a un petit excès de population qui dépend de l’intensité du champ magnétique appliqué.
Les atomes avec I=1/2, tels que 1H et 13C peuvent prendre 2 orientations avec des énergies différentes correspondant au spin +1/2 et -1/2. Le niveau d’énergie inférieure a un petit excès de population qui dépend de l’intensité du champ magnétique appliqué.
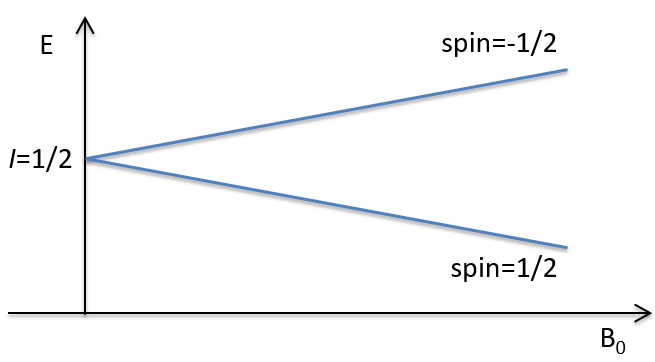
Pour faire la transition entre les deux niveaux, les quanta d’énergie hν, avec ν la fréquence du rayonnement électromagnétique, sont nécessaires dans un champ magnétique B0. La fréquence est liée au champ magnétique par la relation :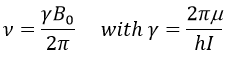
γ est la constante gyromagnétique qui dépend du spin I et du moment magnétique μ. Sachant cela, le problème est de trouver un moyen de transférer cette énergie et de mesurer l’énergie absorbée.
Dans un champ magnétique, un proton tourne sur lui-même et autour de l’axe du champ magnétique (voir la figure suivante).
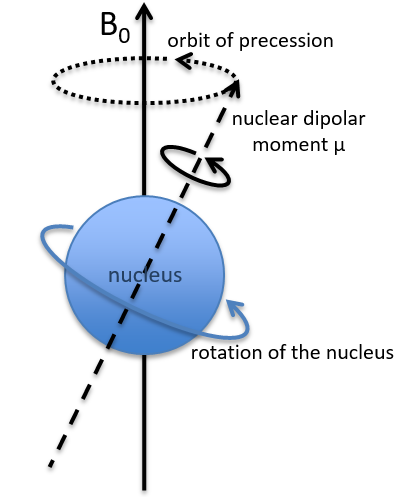
La vitesse de précession ω0, aussi appelée fréquence de Larmor est égale au produit de la constante gyromagnétique γ avec l’intensité du champ magnétique B0.
![]()
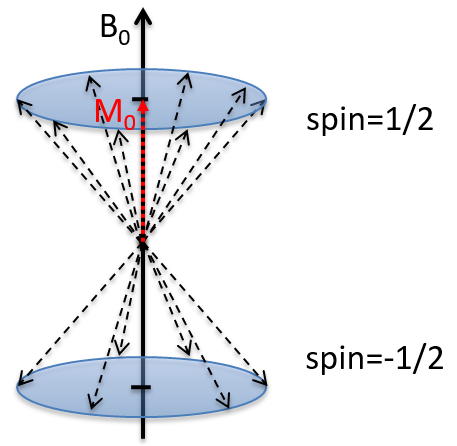
A cause du champ magnétique, tous les atomes sont orientés dans la même direction mais ils ne sont pas au même stade de la précession. Selon le spin, l’orientation est parallèle au champ (spin = 1/2) ou antiparallèle (spin = -1/2). Comme il y a un petit excès de population pour le spin = 1/2, une aimantation globale M0 est dans l’axe de B0 (axe z) mais il n’y a pas de magnétisation particulière dans les autres directions (axes x-y).
Nous inclinerons la magnétisation vers le plan horizontal en utilisant un oscillateur qui génère un second champ magnétique B1 perpendiculaire à B0. Le champ électromagnétique résultant tourne à la même vitesse que le proton. Nous pouvons détecter la composante magnétique générée dans ce plan (My par exemple) si vous le souhaitez. Maintenant que le noyau est excité, nous fermons le champ magnétique B1 et laissons le noyau se détendre jusqu’à sa magnétisation précédente.
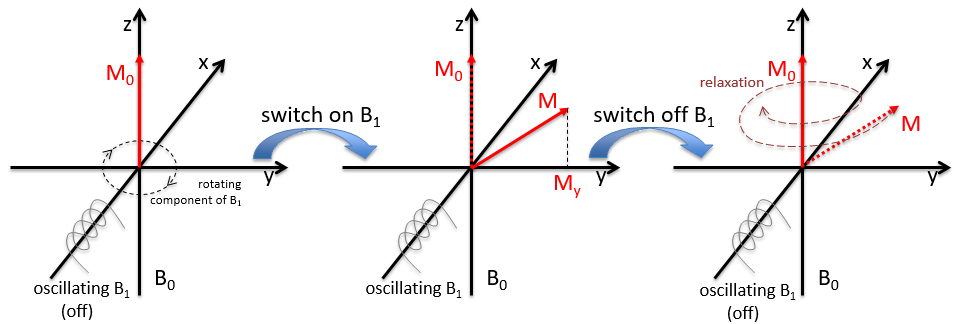
Deux méthodes de relaxation existent. La première est appelée relaxation spin-réseau ou relaxation longitudinale et implique le transfert de l’énergie des noyaux aux molécules voisines. Cette relaxation prend un temps T1. Le deuxième mécanisme de relaxation est la relaxation spin-spin, ou relaxation transversale et implique le transfert d’énergie d’un noyau à un second. Il a un temps caractéristique T2. Nous pouvons définir R1 et R2 comme :
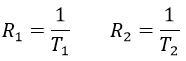
Pour obtenir un spectre de RMN, on scanne soit la fréquence de l’oscillateur B1 soit celle du champ magnétique B0.
Chapitre 6 : Spectres de masse
Le résultat de la spectrométrie de masse est donné sous la forme d’un histogramme donnant l’abondance des espèces en fonction de leur rapport m/z (on l’écrit aussi M, car les ions sont habituellement monochargés). C’est ce qu’on appelle un spectre de masse. L’abondance est donnée en% du plus grand pic. Peu importe le nombre de fragments, il y aura toujours un pic avec une abondance de 100% et les autres pics auront une abondance plus faible.
Les fragments de faible m/z sont sur la gauche et l’ion parent est, finalement, à l’extrémité droite du spectre. L’ion parent est la molécule qui a été ionisée mais n’a généré aucun fragment. Nous pouvons nous référer à son pic comme le pic parent ou le pic M. Son pic n’est pas toujours présent sur le spectre ou il peut être petit. Il est plus commun d’avoir un pic de parent intense dans le cas d’un bombardement chimique pour lequel un pic à M + 1 est également intense. Des pics avec un M plus grand que le substrat peuvent éventuellement exister à cause des isotopes.
La règle de l’azote :
La règle de l’azote est une règle assez simple qui dit que si la masse de l’ion parent est paire, il y a un nombre pair d’azote dans la molécule. Si la masse est impaire, le nombre d’azote est également impair. Par exemple, il n’y a pas d’azote dans C2H6 et la masse est paire: 2×12 + 6 = 30. L’azote peut porter un atome d’hydrogène en moins (NH3) qu’un carbone (CH4). Si l’on remplace l’un des atomes de carbone par un atome d’azote mais qu’il a aussi une masse impaire, on aura CH3-NH2 avec une masse de 12 + 14 + 5 = 31. Si nous étions en présence de NH2-NH2, la masse est paire puisqu’il y a deux atomes d’azote (M = 32).
De plus, le pic M est généralement faible s’il y a de l’azote dans la molécule.
Les pics isotopiques :
C’est assez rare quand un pic n’est pas directement entouré par d’autres pics d’intensités plus faibles. Ces amas sont dus à la présence d’isotopes dans les fragments. L’abondance de 13C est faible: 1,1% par le carbone dans la chaîne mais des pics à M+1 (M étant le rapport m/z sans isotope) peut être observé si l’intensité du pic principal est grande. Il est également possible que deux 13C soient sur le même fragment, conduisant à un très petit pic à M+2. Nous pouvons estimer la quantité de carbone dans le substrat à partir de l’intensité des pics M+1 et M+2. L’intensité du pic M+1 est égale à 1,1% n de l’intensité du pic M, avec n la quantité de carbone (on ajoute également 0,36% pour chaque atome d’azote N dans la molécule). Cependant, des hétéroatomes tels que O, N et S peuvent également augmenter l’intensité du pic M+1. Cette astuce n’est donc pas absolue.
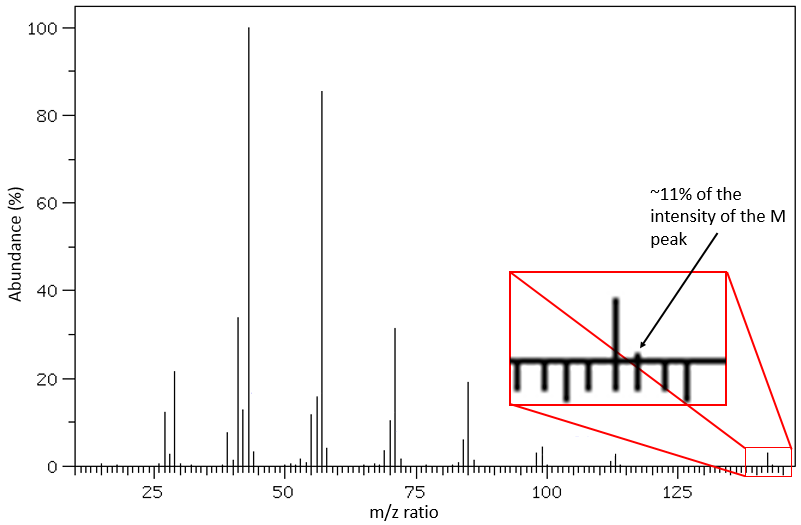
D’autres atomes peuvent facilement être distingués par l’intensité de leurs pics isotopiques à M+2. Cl et Br ont des isotopes à forte abondance. Fragments possédant un de ces atomes peuvent facilement être repérés sur un spectre de masse. S montre également un pic à M+2 d’approximativement 5% du pic M par atome de S. O montre un très petit pic à M+2 (O.2% par atome d’oxygène). Finalement, Si a des isotopes donnant des pics visibles M+1 (5,1%) et M+2 (3,35%).
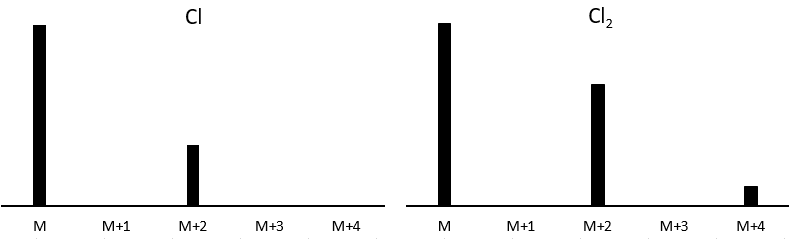
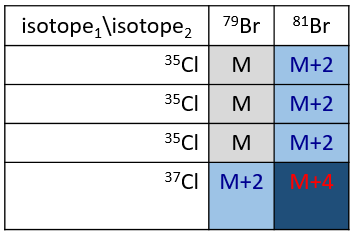
Cl a deux isotopes stables: 35Cl et 37Cl. Leurs abondances sont respectivement de 75% et 25%. Sur le spectre, il en résulte un pic à M+2 avec une intensité correspondant au quart du pic principal. S’il y a deux Cl sur le même fragment, les deux peuvent être 35Cl ou 37Cl ou nous pouvons en avoir un de chaque. Nous aurons donc un pic à m/z = M, M+2 et M+4. L’intensité des pics est toujours liée à l’abondance des isotopes. Pour simplifier, il y a 3 chances sur 4 d’avoir un 35Cl et une chance sur 4 d’avoir un 37Cl pour chacun des Cl. Nous pouvons dessiner le tableau suivant et écrire le pic correspondant dans les cas pour la combinaison correspondante.
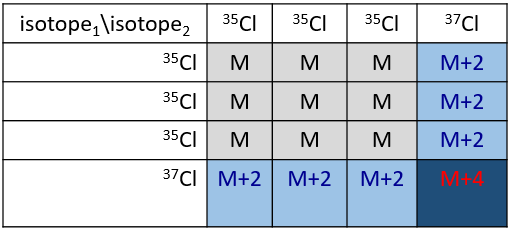
Il y a 9/16 des fragments qui auront la masse M, 6/16 la masse M+2 et 1/16 la masse M+4. Le pic le plus intense est toujours le pic avec deux 35Cl. L’intensité du pic avec un 35Cl et un 37Cl à M+2 représente 2/3 de l’intensité de M et le pic à M+4 avec deux 37Cl un peu plus de 10%.
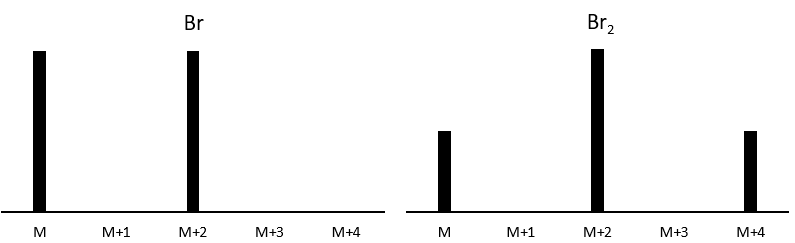
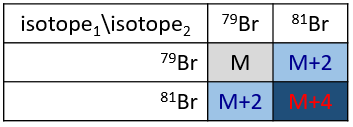
Les abondances des isotopes de Br sont approximativement équivalentes. Si un atome de Br est sur le fragment. Deux pics d’intensités similaires sont séparés par 2 unités de masse. Si deux Br sont sur le fragment, le pic principal est celui qui porte un 79Br et un 81Br (M+2). A M et M+4, les pics ont la moitié de l’intensité de M.
If the fragment has 1 Cl and 1 Br, then we have
The relation between the intensities of isotopic peaks (for one type of isotope) is given by the development of the power of a sum
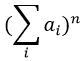
where ai is the abundance of the isotopes i of the atom and n is the number of atoms of the isotope in the fragment. For the example of the bromine, there are two isotopes with equivalent abundances (50%). If there are three atoms of Br in the fragment, we should observe a repartition similar to
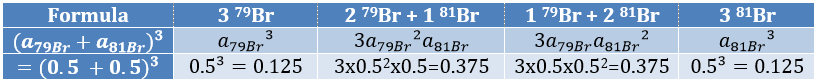
Fragmentation peaks
Now that we have analysed the parent peak, we have some advices on the nature on the substrate but we can only guess which molecule it is. A simple example: how would we know if the substrate is the n-octane of the 2-methylheptane?
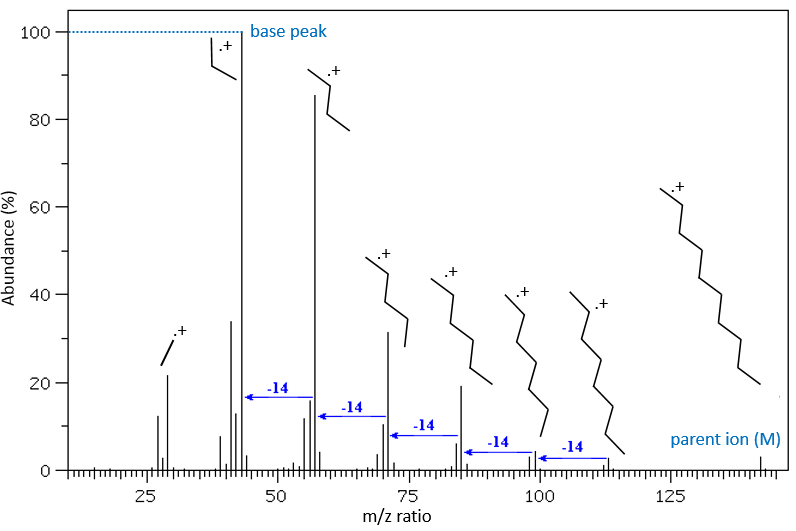
The fragments peaks can help us to find out. To talk about the fragments, we refer to peaks by their m/z ratio. For instance, we can say that the parent ion is at M=m/z=114 and that the base peak is at m/z=43 for both species, and that they have peaks in common (m/z=29, 43, 57, 71 and 114). The intensities of those peaks are not identical and the n-octane shows a peak at m/z=86 and no peak at m/z=99. The fragmentation was thus different because of the structure of the molecule, the composition being identical.
Imagine now that we only knew that the two molecules are alkanes. From the parents ion we can determine that both of them are a form of octane. The interest of the fragmentation peaks will be to give us advices on the structure of the alkanes.
It can be useful to draw the molecules of our guess and cut them into fragments. Virtually, the molecule can break anywhere. A method is to draw the semi-developed representation of the molecule.
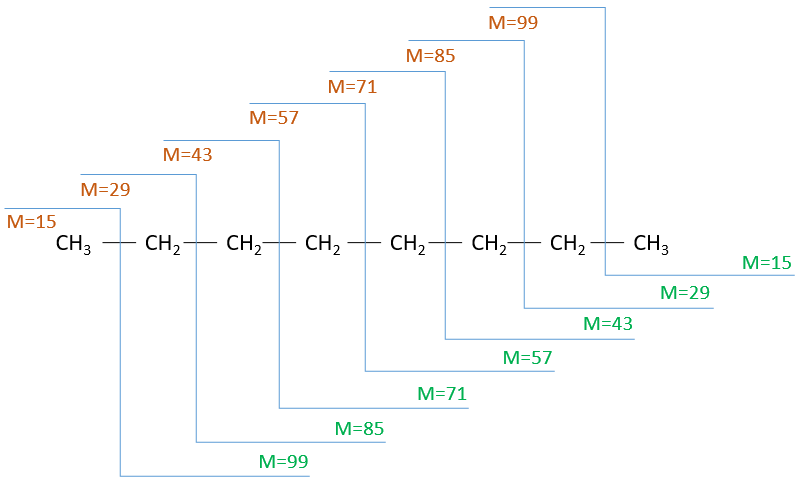
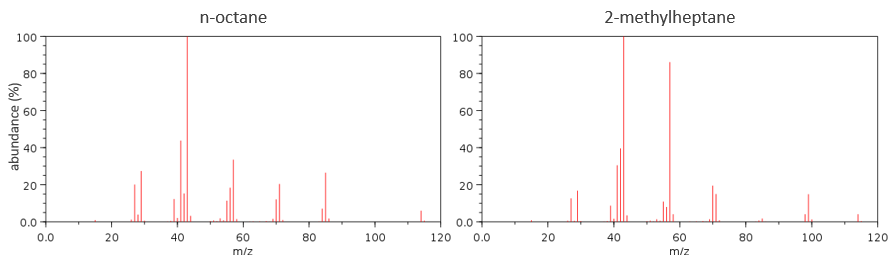
To represent the fragmentation at one liaison, we draw a line perpendicular to the liaison and indicate the masses of the fragments at the left and right of the liaison (respectively in orange and green on the figure above). The Z shape drawn on the figure above is more convenient than a simple line in the case of more complex molecules. For instance, if the n-octane is cleaved after CH3-CH2, the fragment of the left has a mass of 29 (units of mass) and the second fragment, i.e. the rest of the molecule, has a mass of 85. These fragments can be found on the spectrum of the n-octane and have approximatively the same abundance. Unfortunately, the abundances of two complementary fragments are not linked. Remember that when we obtain a fragment, only one side of the molecule remains charged and is detected by the MS, the second fragment being neutral and thus not detected.
![]()
One can see that the fragmentation at the level of the –CH3 does not appear on the spectrum. It is because the probability of fragmentation for a given liaison is related to the strength of the liaison, to the possibility of a low energy transition and to the stability of the generated fragments. In general, the intensity of the peaks are not identical and it is common to observe one or two very intense peaks amongst the fragmentation peaks. Some general rules may help us to predict the nature of the most intense peaks:
- the intensity of the M peak is the highest for linear chains and decreases with the degree of ramification. One can see it on the previous figure.
- the intensity of the M peak decreases with the mass of the substrate (for similar structures). The fat esters are an exception.
- aromatic compounds usually show intense M peaks. Double liaisons, cycles and aromatic cycles stabilise the molecular ion, leading to an intense M peak.
- the fragmentation is favoured at substituted carbons because it leads to carbocations that are more stable. The series for the stability of carbocations is
![]()
The rule is thus similar to the one for the stability of carbocations for SN1. The biggest substituents are usually removed more easily and there is usually no peak at M-15 because CH3+ is not stable. If a peak is present at M-15, it definitively means that there is a ramification in the molecule.
- Double liaisons favour the allylic cleavage to stabilise the carbocation.
![]()
- liaisons nearby heteroatoms are frequently broken because of the possible stabilisation of the charge by resonance.
![]()
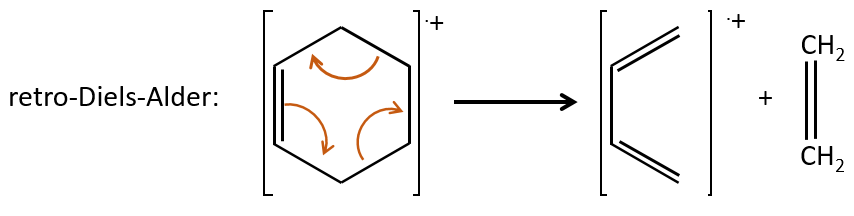
- saturated cycles usually lose alkyl chains and keep the positive charge. Unsaturated cycles are subject to a retro-Diels-Alder reaction.
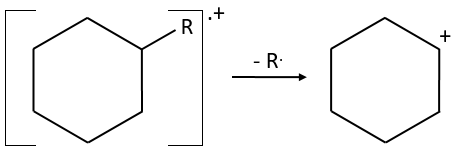
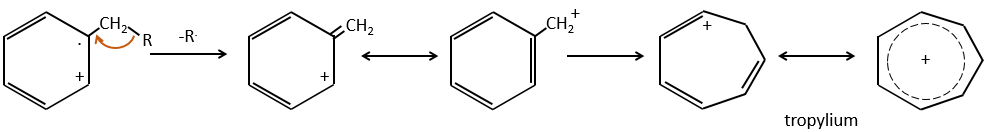
- aromatic cycles usually keep a methyl group and fragment in beta of the cycle to stabilise the positive charge and give the tropylium ion by rearrangement.
- the cleavage is favoured when small neutral molecules can be removed, such as water, H2S, NH3, HCN, CO, etc.
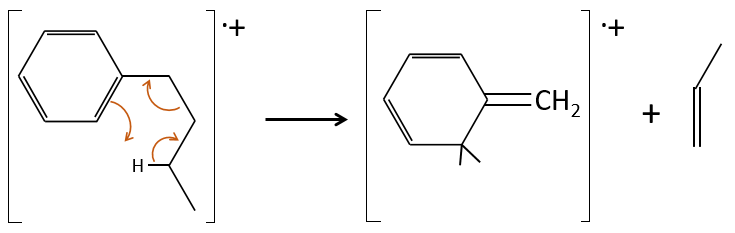
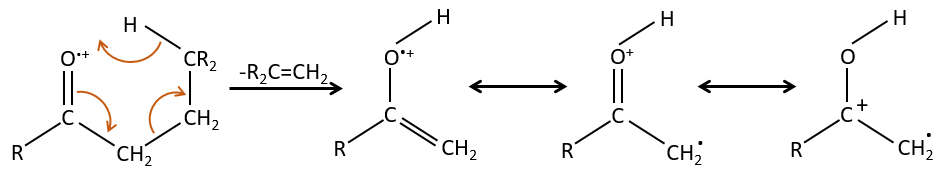
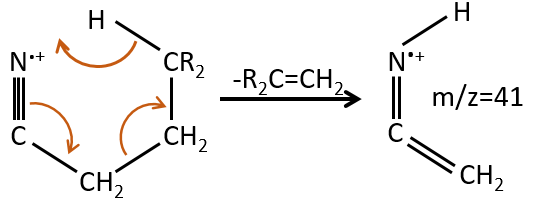
- rearrangement of Mc Lafferty if a hydrogen is in γ of carbonyl. Some fragments cannot be explained be simple cleavages of liaisons from the molecular ion. Such fragments can be the result of an intramolecular rearrangement. The rearrangements often involve the migration of a hydrogen atom on a heteroatom. For instance, a hydrogen in γ of carbonyl can migrate on the O.+. The resulting cleavage is done on the liaison in αβ of carbonyl. It is called a rearrangement of Mc Lafferty.
Typical fragments
There are two ways to look at the fragmentation peaks, and they are used together. The first way is simply to determine what they can be from their m/z, as we have done with the parent peak. There are tables of possible ions corresponding to each m/z ratio.
Here comes a list of some characteristic ionic fragments (that are thus detected on a spectrum):
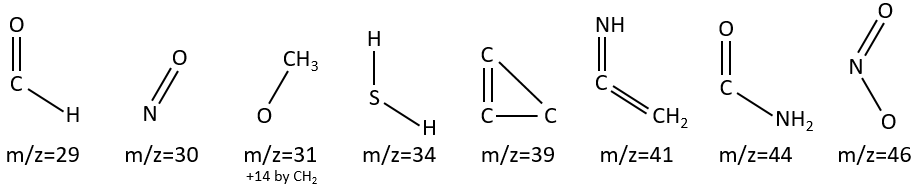
M between 19 and 25: unusual except if there is a O or F.
M=29: presence of an aldehyde.
M=30 (or 46): presence of a nitro.
M=31+n14: presence of an etheric oxygen.
M= 34: presence of a primary -SH.
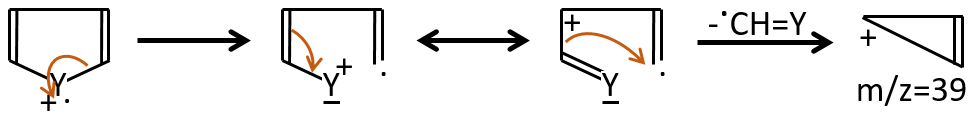
M=39: presence of a heteroaromatic atom in a cycle of 5 atoms. Several complex cleavages can occur in heteroaromatics, giving respectively the ions C3H3+ (m/z=39), HCY+ or CH2Y+.
M=41: presence of a nitrile (if Mc Lafferty).
M=44: presence of a primary amide (if Mc Lafferty).
M=46 (or 30): presence of a nitro.
M=47: presence of a sulfur.
M=59+n14 (clusters): presence of an ester.
M=60: presence of an acid (if Mc Lafferty).
M=60+n14: presence of an aliphatic nitrite.
M=61: primary or secondary -SH.
M=77: this peak is intense and is characteristic of aromatic cycles. It represents the fragment C6H5+. This peak is often in a cluster of peaks (C6H5-6-7) and is accompanied by a peak at M=91 for the toluene/tropylium ion.
M=97: presence of a nitrile (if Mc Lafferty) with a long chain able to form a cycle of 6 carbons.
The other way is to determine which particle has been lost from the parent ion (or from another fragment) to obtain the fragment we are looking at. On the spectrum of the linear alkane that we have seen at the beginning of the section, there is a regular Δm/z=14 between fragments, corresponding each time to the removal of one CH2 fragment of the chain.
It does not mean that the parent ion broke into one fragment by losing one CH2, then that that fragment loses CH2 afterwards, that itself loses CH2 etc., but that the parent ion can break at several places of the chain with a given probability proportional to the intensity of the peak. If the chain is not linear, the distances between the fragments are not so regular. It is why we can say that the spectrum of the left is the one of the n-octane and the second spectrum is the one of the 2-methylheptane.
Here comes a list of some characteristic fragments (neutral or radicals) that can be removed from the molecule:
M-15: -CH3
M-18: -H2O
M-30: -NO
M-31: -OCH3
M-33: -SH
M-45: -COOH
M-46: -NO2
The presence of halogens, spotted by the presence of intense peaks at M+2/M+4 can also be confirmed by the presence of a peak at M-X with X the mass of the halogen.
Confirmation of the composition and structure
Once we determined enough fragments (present on the spectrum and lost), we can try to puzzle them together. At this point, we should have a clear idea of the formula of the molecule. With the formula, we can determine the degree of unsaturation of the molecule. It is given by
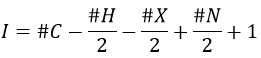
For instance, C7H7NO has a degree of unsaturation I=5. The atoms that are bivalent, such as O, are not counted. The atoms with the same valence than C, H, N and O have the same value of unsaturation. S has a value of 0, P has a value of +1/2, etc.
![]()
One unit of I represents 1 double liaison or one cycle. Triple liaisons are counted as two double liaisons. A phenyl has thus a value of 4 (3 C=C and 1 cycle).
![]()
Y can be a carbon but also a heteroatom. The molecule corresponding to the formula C7H7NO can thus be
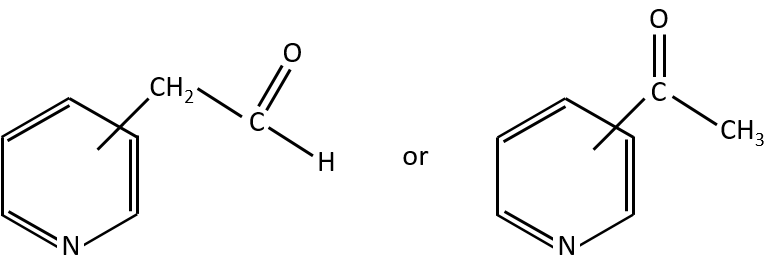
In combination with the mass spectrum, we should be able to determine which molecule it is.
To check our guess, it can be useful to draw the molecule and cut it into its fragments. For instance, the n-butanol and the sec-butanol have two very different mass spectra. Some peaks are on the same location but their intensities are different, referring to the probability of cleavage at a given liaison. It is not necessary to explain all the peaks.
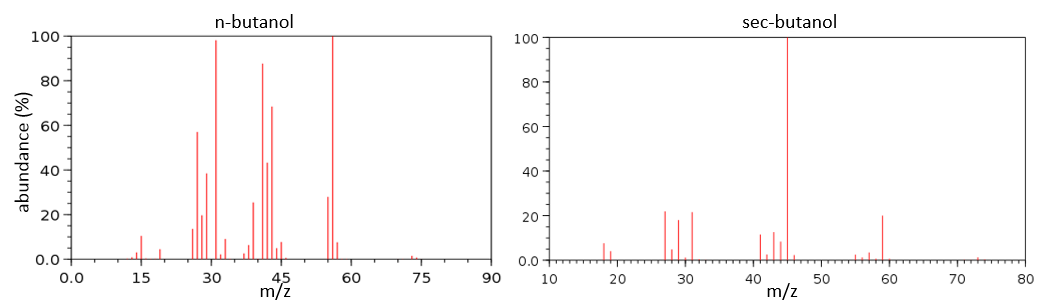
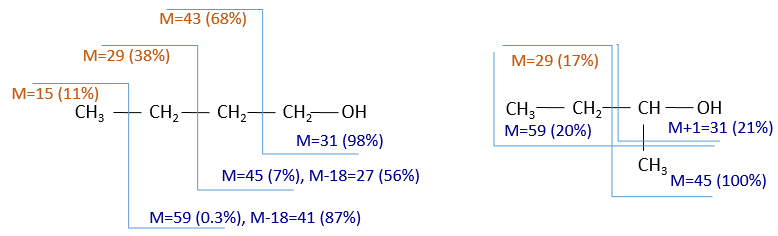
We gave here a strong resume of the mass spectrometry technique and of the analysis of a spectrum. The manual analysis of a spectrum is something that you learn by doing it again and again and that may require some guessing (sometimes some small peaks are important to determine the structure of the molecule and some intense peaks are clueless, like the peak at 56 of the n-butanol). Each group (alcohol, ether, ester, etc) have particular fragments or rearrangements and it is not the point of this section to describe all of them. It will eventually be the subject of a further section (it represents more than 20 pages in my book of reference (Spectrometric identification of organic compounds, Silverstein, Basler, Morill) and the tables for the fragments are 50 pages long, ≈150 fragments per page.
Despite its huge power, there are often several structures than can correspond to one mass spectrum. Usually, the mass spectrometry is used in combination with IR/UV spectrometry and/or NMR. Combined together, we can determine not only the structure of the molecule, but also the exact conformation. We will discuss those techniques in the following sections.
Chapitre 5: Spectrométrie de masse
La spectrométrie de masse est l’une des techniques les plus utilisées au laboratoire car elle permet de déterminer la structure d’une molécule. Pour cette technique, l’échantillon doit être une solution pure, contenant seulement une molécule qui sera analysée dans un délai très court. C’est pourquoi les spectromètres de masse font souvent la queue dans une colonne chromatographique (HPLC ou GC). La solution sortant de la colonne est séparée en petits échantillons directement analysés dans le spectromètre de masse.
Le principe est assez simple: nous détruisons la molécule avec un bombardement d’électrons ou de cations et nous détectons ses bits, les ions gazeux.
Le principe est assez simple: nous détruisons la molécule avec un bombardement d’électrons ou de cations et nous détectons ses bits, les ions gazeux.
Les clivages des molécules ne sont pas totalement aléatoires et obéissent à quelques règles. Il en résulte des ions caractéristiques dont la nature peut être directement déterminée. La détection des ions se fait en fonction de leur rapport masse/charge m/z (et donc de leur masse). La masse des ions est l’addition de la masse des atomes qui les composent. Notez que la masse inscrite sur la table de Mendeleev (la masse chimique) n’est pas la masse d’un atome mais l’addition pondérée des masses de ses isotopes. L’utilisation des masses chimiques est pertinente dans le cas de solutions macroscopiques, mais dans le cas de la spectrométrie de masse, nous détectons chaque ion unique de sorte que les masses atomiques doivent être utilisées. Le fait que des isotopes différents d’un seul atome soient dans l’échantillon peut même nous donner des conseils sur la nature de l’espèce ionique. Pour découvrir quelle molécule était dans l’échantillon, nous avons assemblé les fragments. Il y a quelques programmes qui peuvent faire le puzzle par lui-même.
Le spectromètre de masse :
Le principe d’un spectromètre de masse est de dévier ou d’accélérer les ions depuis ou vers un détecteur en fonction de leurs masses. Plusieurs modèles de SEP existent et ce n’est pas le but du cours de les décrire tous mais il est toujours important de comprendre comment fonctionne la SEP. Un spectromètre est toujours composé de quelques compartiments caractéristiques:
a) une chambre d’ionisation
b) un séparateur / analyseur
c) un détecteur
Voici un diagramme d’un spectromètre de masse avec déviation magnétique :
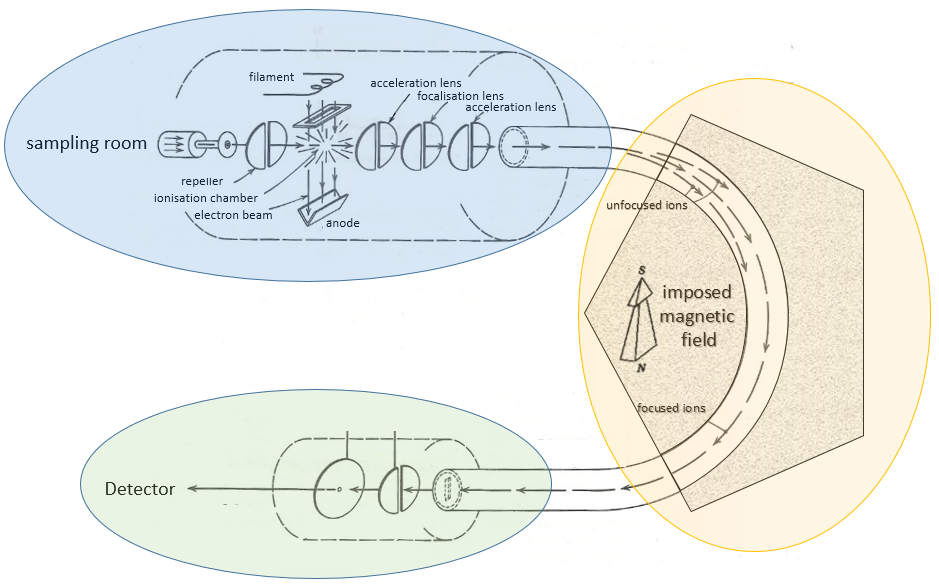
Nous allons décrire brièvement chaque partie.
Échantillonnage
La première étape de l’analyse consiste à ioniser l’échantillon. Cela se fait dans une chambre d’ionisation. Il y a trois façons d’introduire un échantillon dans cette chambre:
- Système d’introduction directe: l’échantillon est placé dans un capillaire placé dans la chambre d’ionisation. Dans cette chambre, la pression est extrêmement faible (≈10-5 torr): on s’attend à ce que le vide évite toute recombinaison entre les ions. La différence de pression transportera l’échantillon là où il sera ionisé. Cette méthode est utilisée pour les échantillons qui ne sont pas volatils.
- système de réservoir: l’échantillon est injecté dans un four à réservoir chauffé à une température donnée. Les échantillons volatils deviennent gazeux et sont transportés vers la chambre d’ionisation par une différence de pression. La pression dans le réservoir est fixée à 10-2 torr contre 10-5 torr dans la chambre.
- par chromatographie: un échantillon gazeux provient directement d’un chromatographe en phase gazeuse (CG). Avant l’introduction dans la chambre d’ionisation, nous séparons le substrat du gaz porteur. La masse de gaz porteur est en général inférieure à la masse du substrat. Nous pouvons utiliser cette différence pour les séparer: un mince tube est devant la sortie du CG. Une pompe est placée perpendiculairement pour pomper le gaz. Comme le gaz porteur est plus léger que le substrat, il est plus dévié et n’atteint pas le tube de sortie. L’échantillon est ainsi purifié.
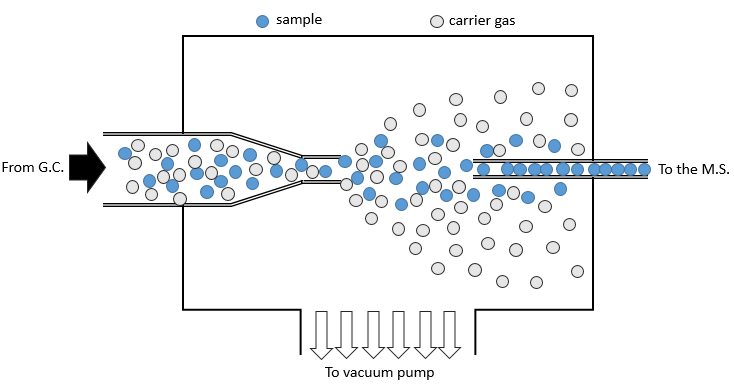
Ionisation :
Le but de l’ionisation est de former des ions chargés positivement à partir du substrat. Pour ce faire, l’échantillon est bombardé (par des électrons ou par des cations) pour expulser plus d’électrons des molécules
![]()
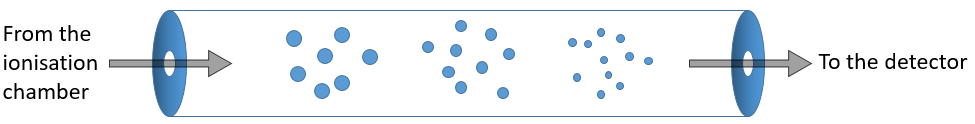
La plage d’énergie typique requise pour l’ionisation est de 8 à 15 eV. Comme nous voulons être sûrs que le substrat est ionisé, nous injectons plus d’énergie que nécessaire (50-70eV). Il est possible d’obtenir une double ionisation. L’excès d’énergie peut briser l’ion en ions plus petits.
![]()
Ce processus peut être répété plusieurs fois et à partir d’un substrat, nous pouvons avoir des dizaines de fragments.
Il y a deux méthodes d’ionisation:
- bombardement électronique
Un faisceau d’électrons provient d’un métal chauffé tel qu’un filament de tungstène. Les électrons entrent en collision perpendiculairement avec le substrat pour produire les espèces ionisées qui sont dirigées vers le détecteur par un répulseur. Les molécules neutres (non ionisées ou résultant des clivages subséquents) sont éliminées par une pompe - bombardement chimique
Cette méthode est plus douce que le bombardement électronique. Cela conduit à moins de fragmentation. L’énergie d’ionisation est donnée par la collision avec des cations qui finissent par donner un proton au substrat, augmentant sa masse d’une unité (c’est important pour plus tard).
L’analyseur
La différence entre les différents modèles de spectromètres de masse réside essentiellement dans la méthode de séparation dont ils sont équipés.
Les espèces ionisées ne sont pas directement envoyées au détecteur. Avant cela, ils doivent passer par un analyseur. Son rôle est de savoir s’il faut bloquer certaines espèces avant le détecteur ou les séparer sur leur chemin.
Temps de transist :
les molécules ionisées sont accélérées directement vers le détecteur sans aucune déviation. Le temps entre l’ionisation et la détection est mesuré. Ce temps est caractéristique de la masse de la molécule (t~√M): les petites molécules font la distance dans un temps plus court que les grosses molécules.
Déviation magnétique :
Sur le chemin du détecteur, un aimant électromagnétique est mis en route vers le détecteur, déviant les molécules d’un certain angle en fonction de la masse de la molécule.
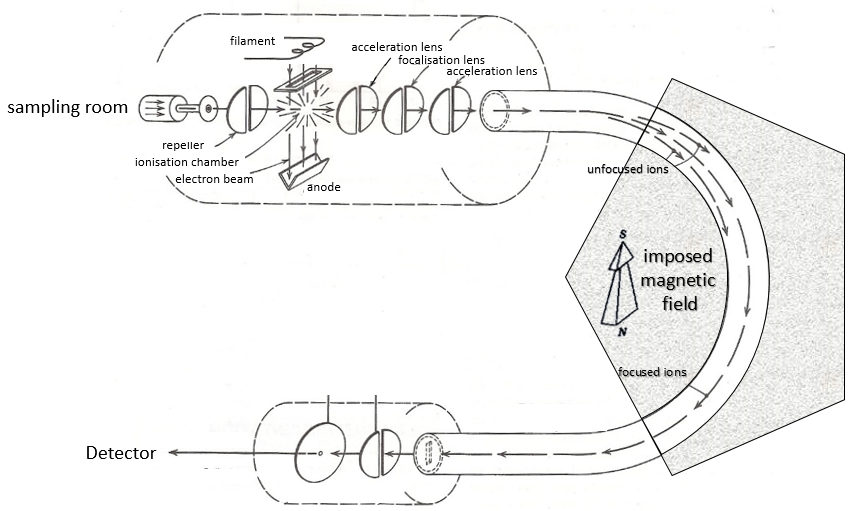
L’énergie cinétique Ek des ions est donnée par la répulsion du répulseur et la vitesse v des ions dépend donc de leur masse m et de leur charge z.
L’énergie cinétique Ek des ions est donnée par la répulsion du répulseur et la vitesse v des ions dépend donc de leur masse m et de leur charge z.
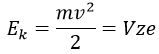
V est la différence de potentiel entre les lentilles d’accélération. Pour atteindre le détecteur, les ions traversent l’aimant et sont déviés par son champ magnétique B. Une seule trajectoire mène au détecteur, lorsque la force centrifuge est égale à la force centripète magnétique, c’est-à-dire quand :
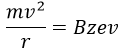
On peut ainsi déterminer le rapport m/z des ions qui atteignent le détecteur en fonction du champ magnétique appliqué B:
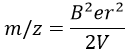
The same process can be repeated with a second magnets, in the case of double focalisation spectrometers.
Quadrupole
On their way to the detector, the ionised molecules are surrounded by four long parallel electromagnets.

Parallel magnets are set to one given frequency. It allows to control the trajectory of the molecules with one specific mass/charge ratio. Only those species will have a stable path that is parallel to the magnets and reach the detector. The other species are deflected and will eventually collide with one of the magnet where they lose their charge. A range of frequencies is scanned to allow all of the ionised molecules to reach the detector separately.
Ion trap
The principle is similar to the one of the quadrupole except that the ions are maintained in a room between electrodes. They are not moving towards the detector. The ions which are in resonance with the electric field remain trapped. If we increase the tension, the ions of larger mass are stabilised while the trajectory of the lighter ions becomes instable and they hit the electrodes.
Detector
As we detect single ions, the signal has to be magnified several times. It is done by a series of dynodes that increase the signal first obtained on a cathode. The final signal reaches an anode and has been multiplied by 106-7 on its way.
The accuracy of the setup has not to be extremely high and it would actually change almost nothing. The important is that a difference of 1 unit of atomic mass can be detected.
Chapitre 4 : Chimie organique – Exercices
Cette section fait partie intégrante du cours et contient quelques réactions qui n’ont pas été abordées dans le cours principal. Il est difficile de comprendre par vous-même certains mécanismes avec les conseils qui sont dans les exercices. Si vous ne réussissez pas, ne vous inquiétez pas. Les solutions et la description des réactions suivent directement chaque exercice. Cependant, les parties qui ont été vues dans le cours sont considérées comme connues et ne peuvent pas être expliquées en détail.
Le principe des exercices suivants est assez simple (mais les exercices ne le sont pas): Nous vous donnons une série de réactions qui se suivent mais nous ne vous donnerons que quelques-uns des réactifs et/ou des produits. De là, vous devez reconstruire tout le processus et découvrir quelle molécule et quelle structure se cachent derrière cette lettre.
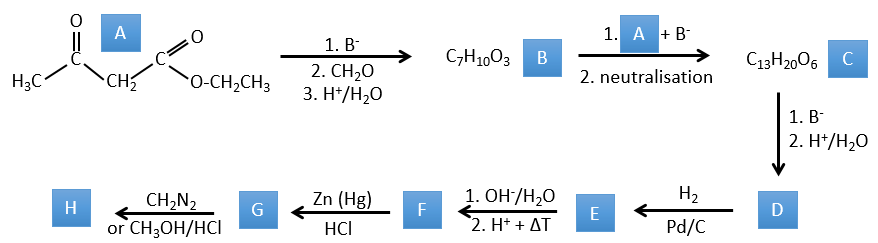
1. Dans ce premier exercice, vous avez la structure du réactif initial (A) et la composition du premier produit B. Vous devez déterminer la structure de B, le produit obtenu par la première réaction. Ensuite, à partir de B, vous devez déterminer la structure de C, puis le composé D et sa structure, etc.
Correction
A → B: Deux ensembles de protons sont plus acides que les autres: les protons en α de carbonyle. Les protons de CH3 sont moins acides que le CH2 parce que CH2 est entre deux carbonyles. C’est là que la base attaque. Le carbone négatif attaque ensuite le formaldéhyde. Un réarrangement se produit après la neutralisation pour obtenir B et H2O.
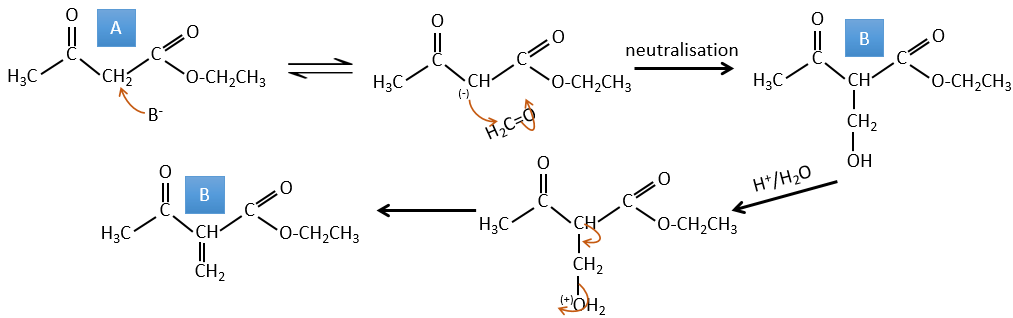
B → C: La première étape est identique à celle de la réaction précédente et les deux molécules se confondent. L’attaque se fait sur le carbone sp2 et on obtient une structure stabilisée par une liaison H formant un cycle entre l’OH et le C = O.
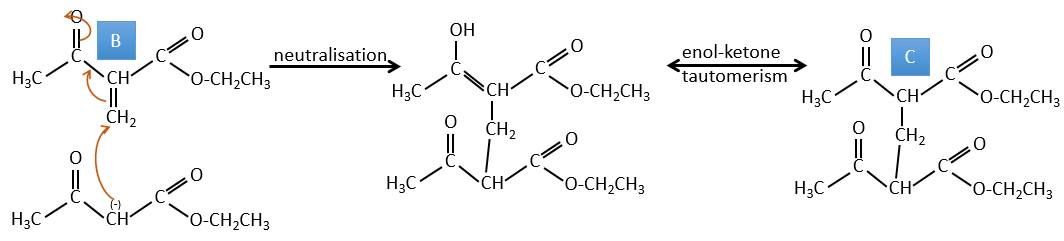
C → D: Cette fois, le proton (noté *) n’est pas attaqué par la base parce que l’élimination d’un proton méthylique permet la formation d’un cycle de 6 carbones. Dans la seconde étape, l’acide catalyse la perte d’une molécule d’eau pour obtenir une double liaison conjuguée au carbonyle.
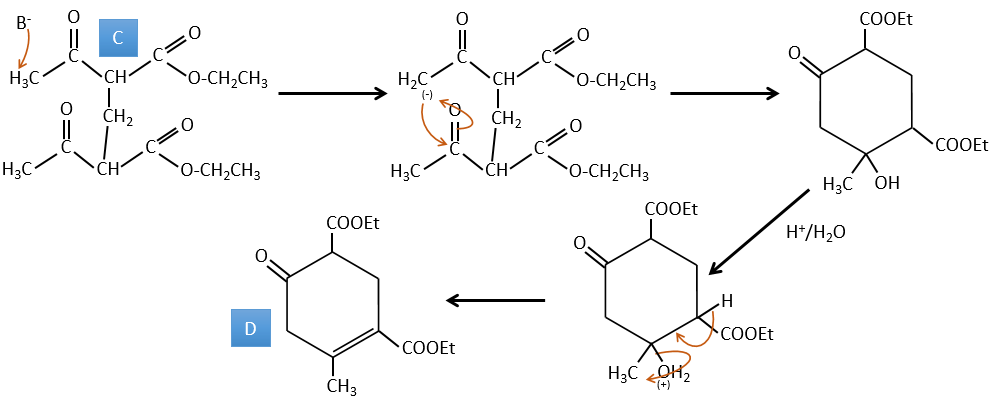
D → E: La double liaison est réduite pour obtenir un cyclohexane.
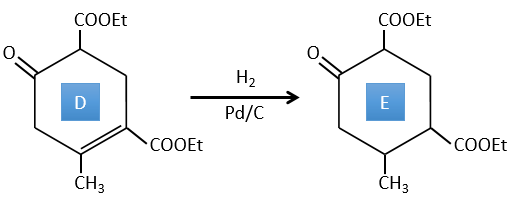
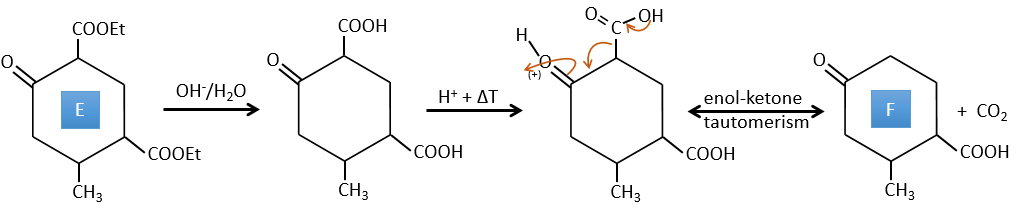
E→F:Les esters sont transformés en acides carboxyliques avec une catalyse basique. Le CO2 se détachera si on augmente la température.
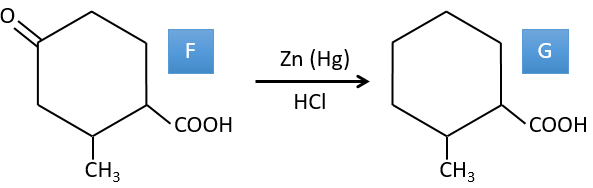
F→G: le Zn réduit sélectivement une cétone dans une chaîne alkyle.
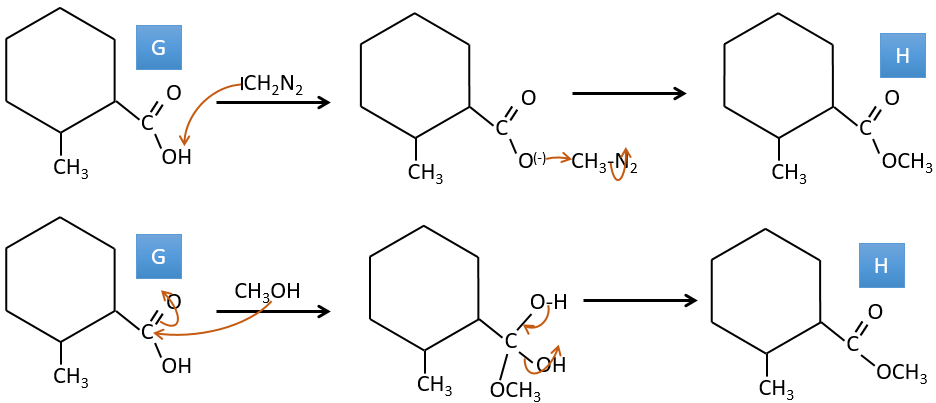
G→H: Le diazonium carbène est capable de prendre le proton de l’acide. Le carboxylate attaque ensuite le carbène pour former un ester méthylique et libérer N2. CH3OH conduit au même résultat mais le processus est différent: il y a une substitution nucléophile sur le carbonyle pour remplacer -OH par
-OCH3.
2. Cet exercice met l’accent sur les réactions concernant les produits aromatiques et les acides carboxyliques ainsi que leurs dérivés.
Correction
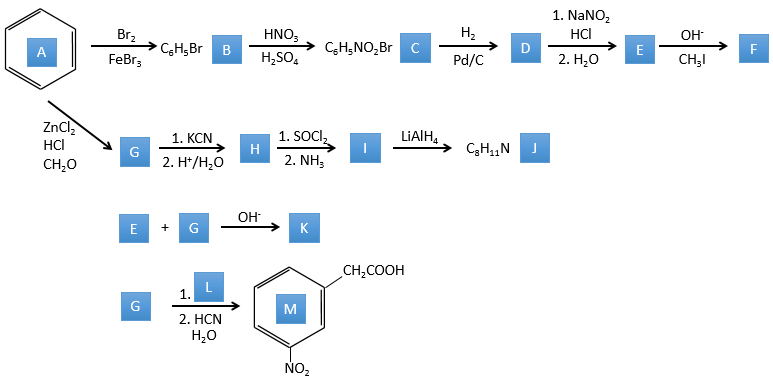
A→B: Le caractère électrophile de Br doit être renforcé par un acide de Lewis pour effectuer la réaction.
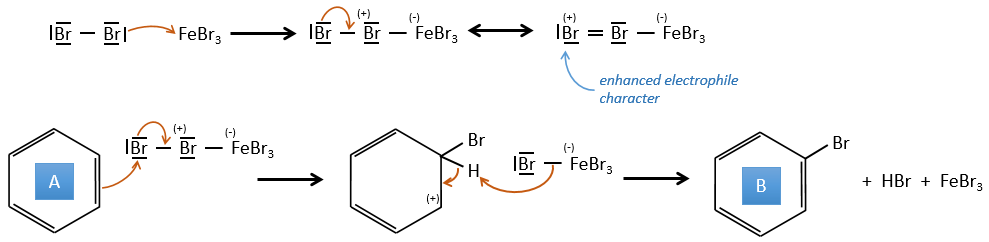
B→C: Un groupe nitro est ajouté au cycle. Il ya déjà un substituant sur le cycle donc nous devons déterminer si le groupe nitro est ajouté en ortho, méta ou para. Un halogène oriente la réaction sur les positions ortho / para. La position para doit être favorisée en raison de l’empêchement stérique sur la position ortho.
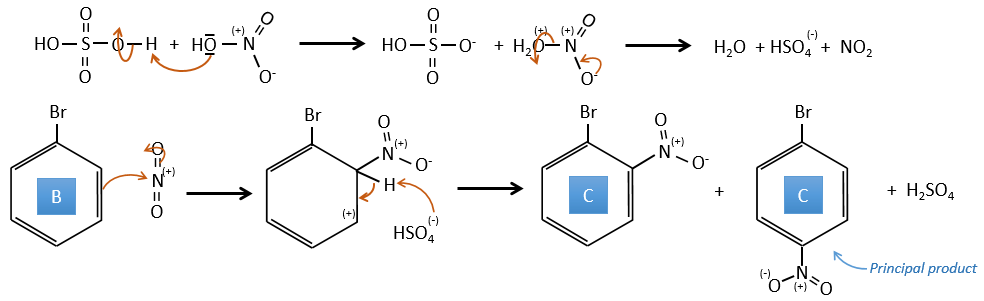
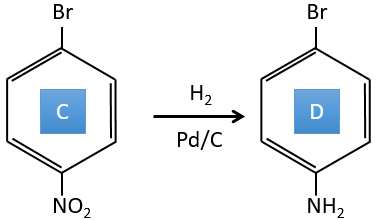
C→D: La réduction est limitée au groupe nitro qui se transforme en amine. Pour réduire complètement le cycle aromatique, nous devons chauffer la solution à 300 ° C.
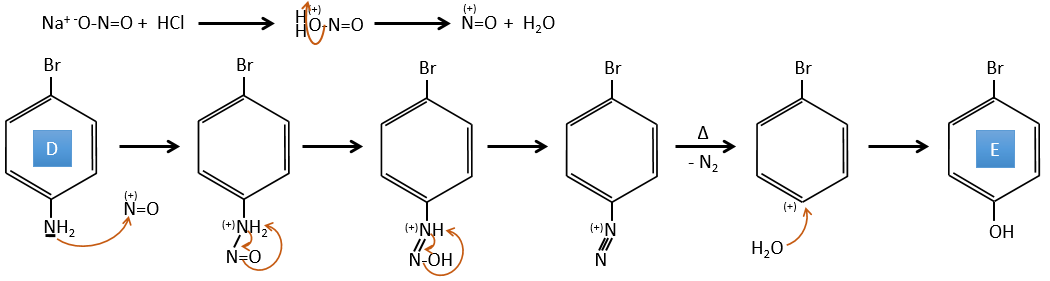
D→E: NaNO2 n’ajoute pas un groupe nitro sur l’anneau. Il conduit à l’élimination de l’aminé et à la formation d’un arénium. Cette espèce très réactive réagit avec l’eau pour remplacer l’aminé par un groupe hydroxyle -OH.
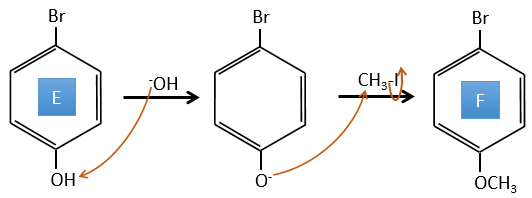
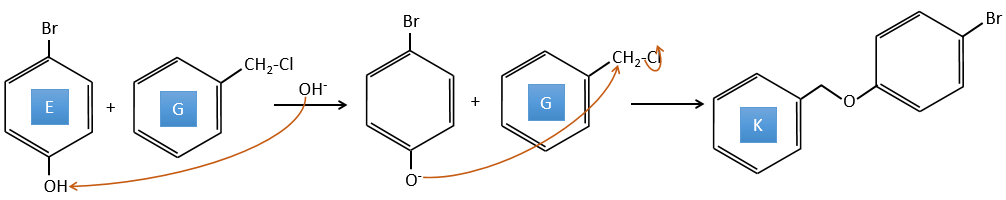
E→F: La base prend le proton du p-bromophénol. Il y a alors une attaque nucléophile par l’anion sur CH3I pour obtenir le p-bromométhoxybenzène.
A→G: C’est la réaction de chlorométhylation. Au cours de cette réaction, le formaldéhyde et l’acide chlorydrique forment un chlorométhanol stabilisé par ZnCl2. L’acide protonne l’alcool et l’anneau peut l’attaquer pour rejeter l’eau et lier le CH2Cl.
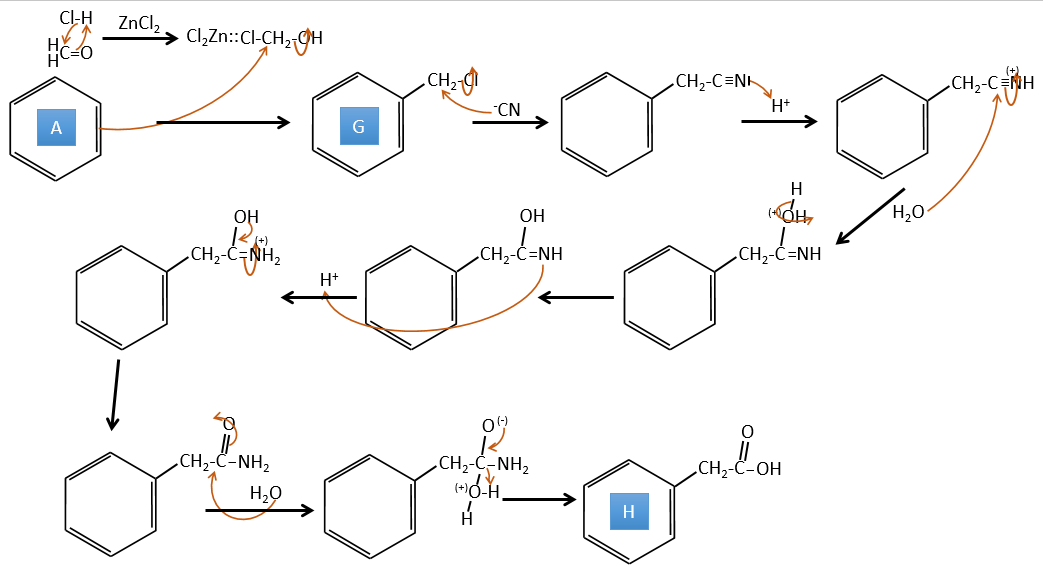
G→H:Un simple SN2 par CN- suivi de sa transformation en un acide carboxylique. Cette transformation se fait par des attaques successives de molécules d’eau sur la liaison carbone à l’azote.
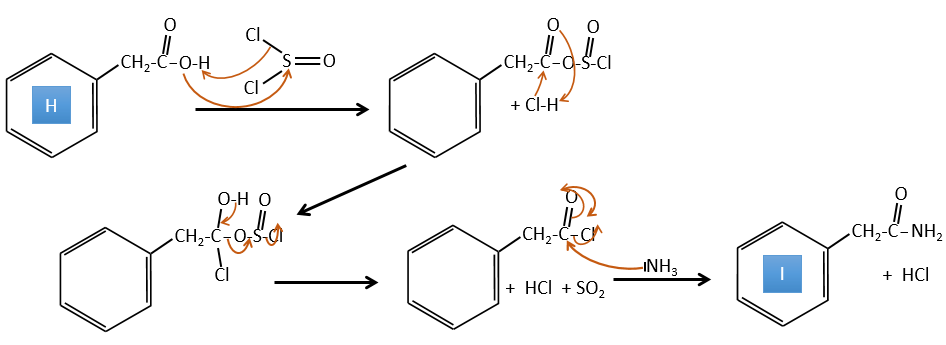
H→I: SOCl2 est une molécule qui nous permet d’obtenir un chlorure d’acyle à partir d’un acide. Cela ne peut pas être fait avec HCl ou Cl2 car Cl- est un meilleur groupe partant que OH-. La réaction est suivie de la formation d’un amide primaire.
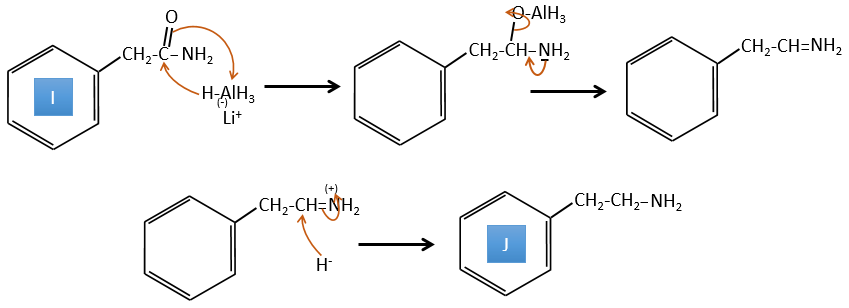
I→J:L’amide est réduit en une amine par LiAlH4. LiAlH4 peut générer H- qui attaque le carbonyle.
E+G→K: Une base prend le proton du bromophénol pour obtenir un nucléophile plus fort. IN SN2 a lieu entre les deux espèces pour les fusionner en une seule molécule.
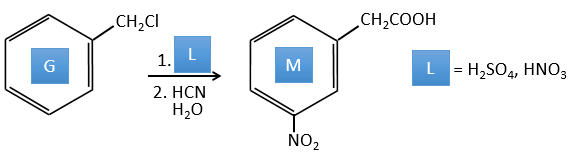
G→M: La seconde étape de la réaction conduit à la formation de l’acide carboxylique comme c’était le cas dans la réaction G-> H. L’élément manquant sur M est le groupe nitro en méta. Cette position est favorisée en raison de l’effet de capteur mésomère du COOH à travers le CH2. L’effet est toutefois plus faible que pour un capteur mésomère directement en contact avec le cycle aromatique.
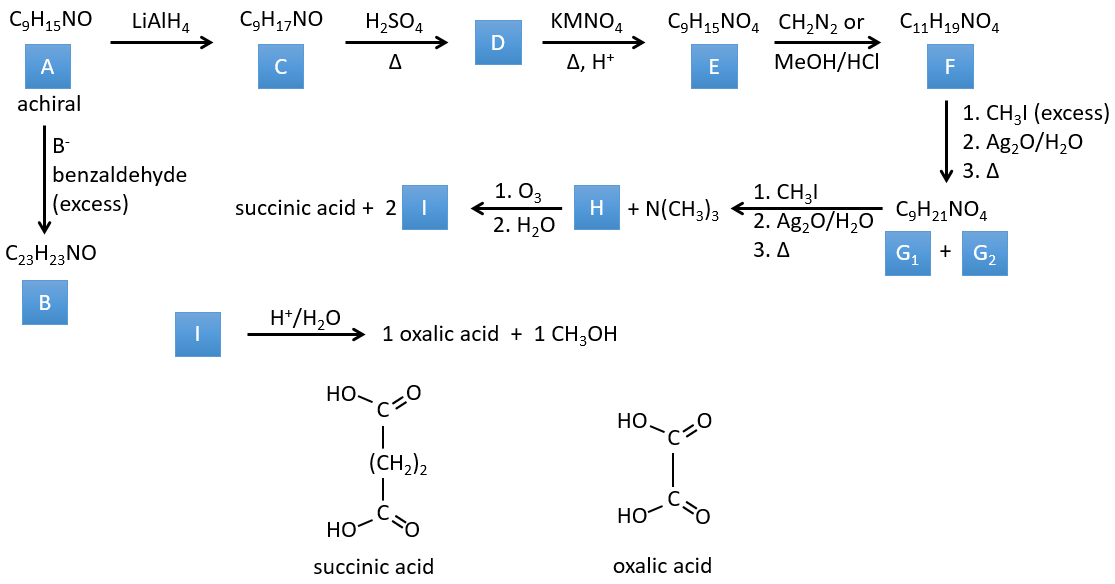
3. Dans cet exercice, le dernier produit d’une longue série de réactions est donné. C’est le produit direct d’une réaction d’ozonolyse. Vous devez donc revenir en arrière dans les réactions, à partir de la fin pour trouver les réactifs de chaque réaction. Les formules de la plupart des molécules sont données. G1 et G2 sont des isomères.
Correction
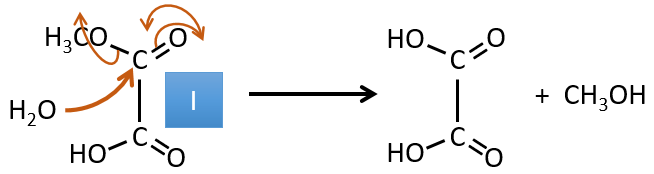
I→…:L’un des produits, l’acide oxalique, est un acide carboxylique et un réactif est l’eau. On peut donc deviner que la réaction est une réaction de substitution sur un dérivé de l’acide carboxylique. L’autre produit de la réaction est une molécule de méthanol. Un acide carboxylique était donc un ester avant la réaction. Un seul méthanol est généré par la réaction, de sorte qu’un seul des deux acides de l’acide oxalique était un ester.
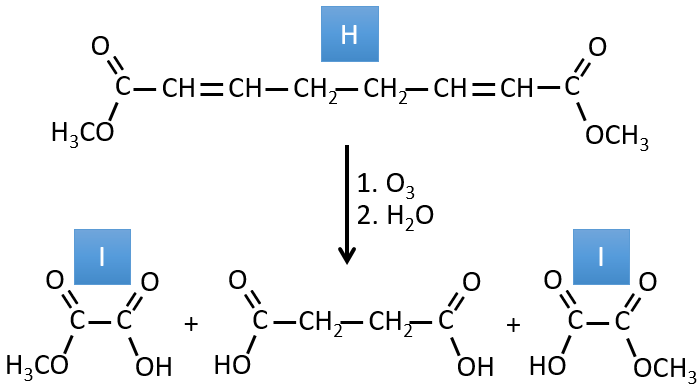
H→I: L’ozonolyse coupe une molécule à double liaison et conduit à la formation d’acides carboxyliques en présence d’un oxydant. Il y a 4 groupes acides dans les produits de cette réaction et 2 esters. Les 4 groupes acides indiquent qu’une plus grande molécule a été coupée à deux endroits. Les doubles liaisons ont été ainsi conjuguées avec les esters. C’est donc un exemple de réaction qui implique une partie du système conjugué et pas tout. Nous ne savons pas si les doubles liaisons sont cis ou trans.
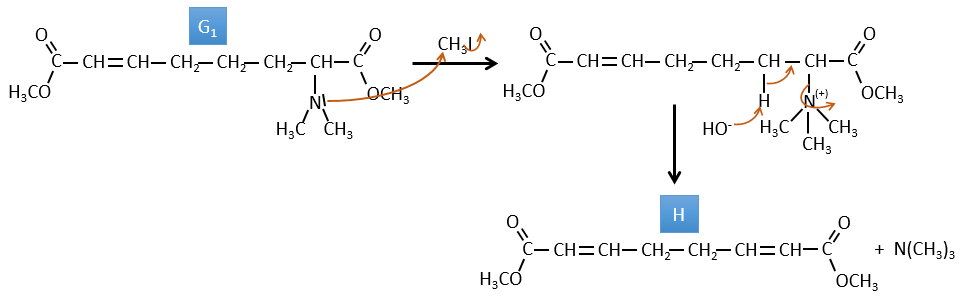
G→H: G1 et G2 sont deux isomères. Il y a deux autres informations que nous pouvons considérer pour trouver les isomères. CH3I, Ag2O et delta sont les réactifs de la réaction de Hoffmann. Cette réaction brise une liaison C-N et forme une double liaison sur ce carbone. C’est donc l’une des liaisons pi que l’azote était liée. La deuxième information est que l’azote n’est plus sur le produit, ce qui signifie qu’il n’avait qu’une seule liaison avec la chaîne. Le deuxième produit de la réaction, N(CH3)3N, le confirme. Les deux isomères sont donc différents du carbone sur lequel N(CH3)2 était lié. Il peut s’agir du carbone en α ou en β du carbonyle.
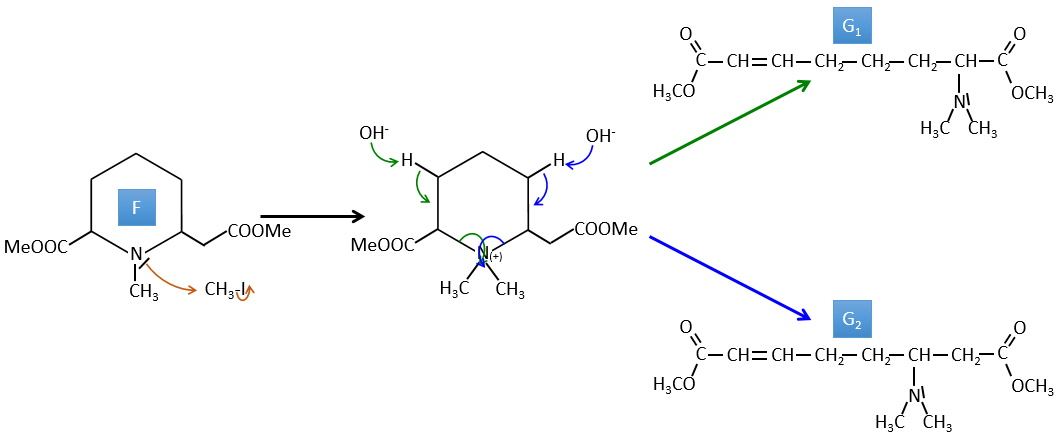
F→G: C’est la même réaction que G-> H mais l’azote est toujours sur la molécule après la réaction. Cela signifie qu’il était lié ailleurs sur la molécule. L’emplacement est l’endroit où se trouve la liaison pi. La molécule avait donc un cycle de 6 atomes avant la réaction. Contrairement à l’espèce H, le cycle F n’est pas symétrique. C’est pourquoi nous pouvons obtenir deux isomères G1 et G2.
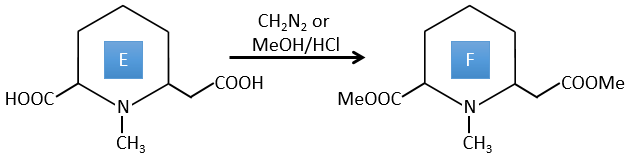
E→F: CH2N2 and MeOH/HCl sont deux techniques pour remplacer un acide carboxylique par un ester méthylique. L’espèce E a donc deux acides carboxyliques.
D→E: KMnO4 agit comme l’ozone. Les deux acides carboxyliques forment ainsi un pont du cycle. Ce pont forme également un cycle de 6 atomes du côté de l’azote (et un cycle de 8 carbones avec l’autre côté).
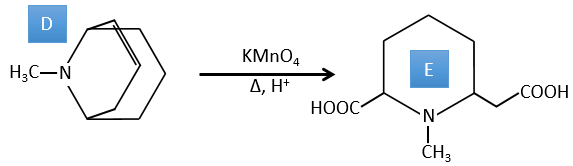
C→D: Si l’on vérifie les compositions du réactif C et du produit D, on voit qu’il y a une différence de H2O. Le rôle de H2SO4 était donc d’éliminer cette molécule d’eau de C avec la formation d’une double liaison. Le groupe hydroxyle pourrait être à deux endroits (dans α ou β des carbones pontés). À ce stade, nous ne pouvons pas dire quelle est la position correcte, mais la réaction A-> B n’est possible qu’avec le groupe hydroxyle dans β du carbone ponté. L’espèce est donc symétrique et achirale comme l’espèce A.
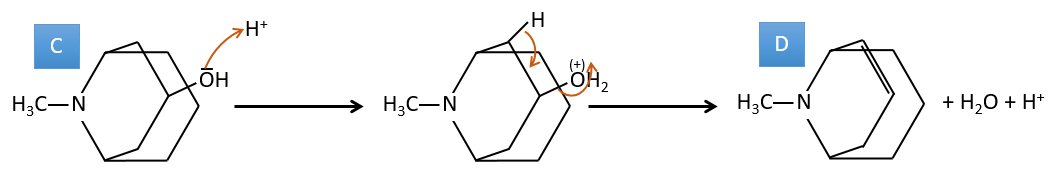
A→C: Une réaction classique de réduction. A obtient H2 dans le processus et nous pouvons supposer que le groupe hydroxyle était une cétone avant la réduction.
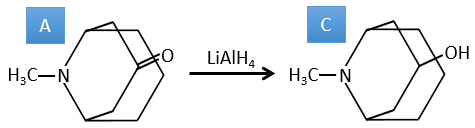
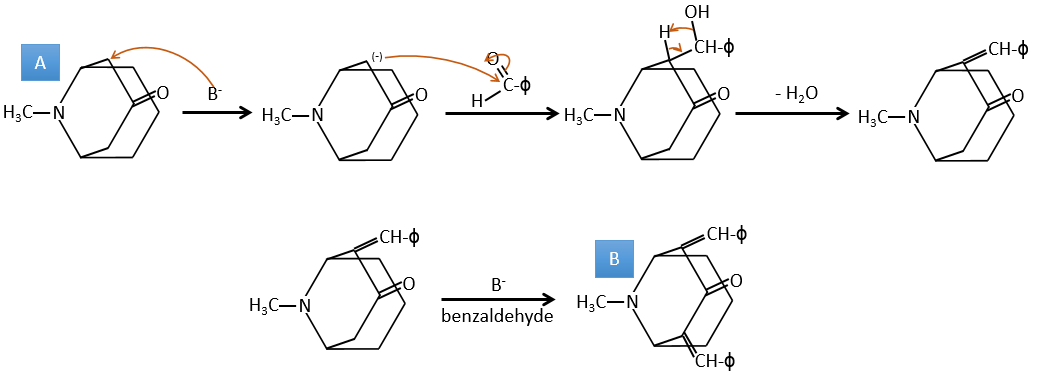
A→B: La base est là pour retirer un proton en α de carbonyle. Ces protons sont acides en raison de la tautomérie énol-cétone. Le carbanion attaque un benzaldéhyde sur son carbonyle et l’eau est perdue après cette attaque. La double liaison est en α de carbonyle et forme une longue chaîne de résonance avec le phényle. Cette réaction peut être répétée de l’autre côté du carbonyle pour obtenir le produit B.
4. Il y a quelques réactions spécifiques dans cet exercice (principalement BàE). Vous avez les formules de tous les composés mais seulement la structure du composé K pour commencer. HNO3, ΔT est un réactif qui rompt la liaison C-C d’une cétone pour obtenir deux acides carboxyliques. Le mécanisme est inconnu.
Correction
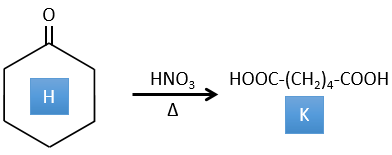
H→K: Comme expliqué dans le libellé, HNO3 casse une cétone en deux acides. Comme les deux acides sont présents dans le produit, H est un cycle avec une cétone. Le cycle a 6 carbones.
H→I: Cette réaction conduit à la formation d’un oxime, c’est-à-dire une base de Schiff où R = OH. Le mécanisme implique une substitution nucléophile par l’azote sur le carbonyle pour rejeter une molécule d’eau.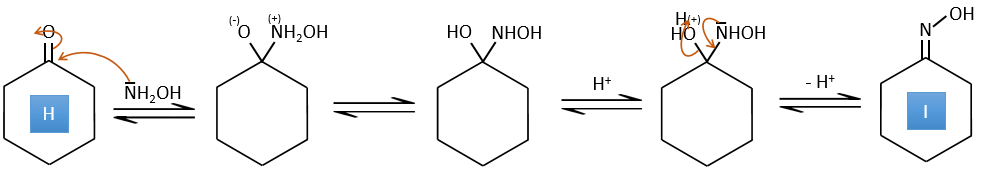
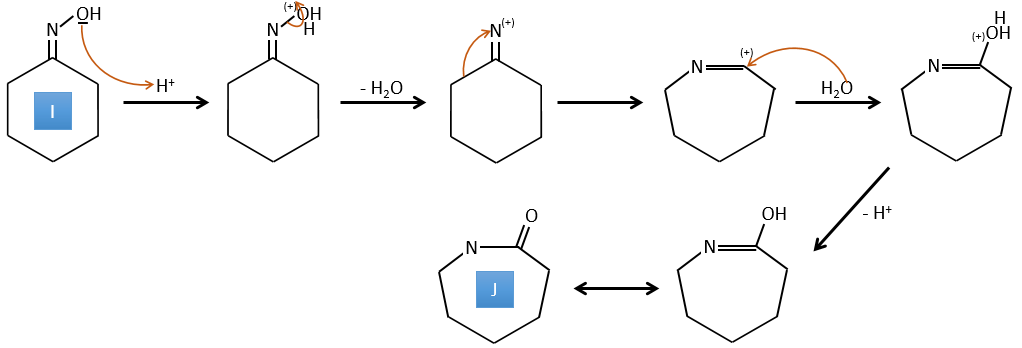
I→J: Cette réaction est la transposition de Beckmann. Dans des conditions acides, le OH de l’oxime est protoné et l’eau est libérée. L’azote cationique est ensuite attaqué par un carbone en alpha de l’oxime, plaçant l’azote dans la chaîne. L’eau revient attaquer le carbocation et former un amide.
G→H: Une réaction simple pour changer un ester en acide carboxylique. Le groupe est ensuite retiré de la molécule par une élévation de température.
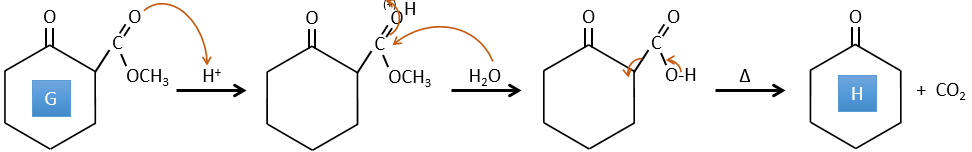
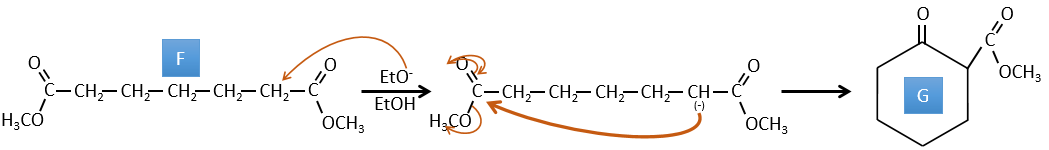
F→G: Un proton en α sur carbonyle est pris par la base forte. L’anion formé attaque ensuite l’ester pour former un cycle de 6 carbones portant une cétone et un ester méthylique.
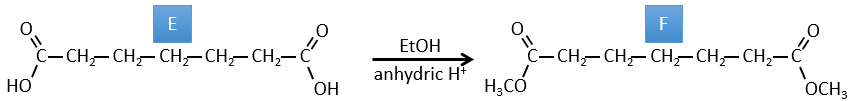
E→F: Les acides carboxyliques sont remplacés par des esters méthyliques
D→E: Ag2O est capable d’oxyder un aldéhyde en un acide carboxylique.
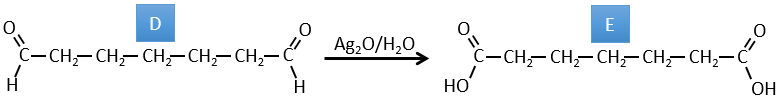
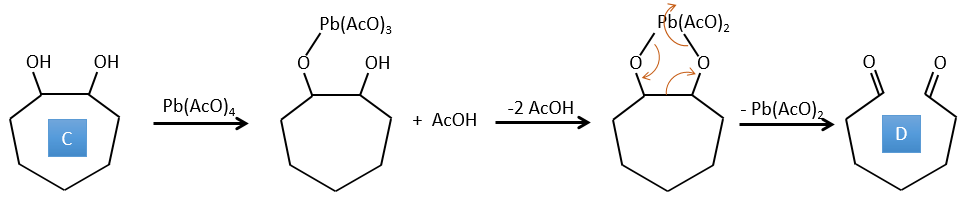
C→D: Le Pb(AcO)4 est un composé qui réagit avec les cis-glycols. Le Pb échange un équivalent d’acide acétique pour se lier à un oxygène. Le processus est lent mais il se liera également avec le second OH pour former un cycle de 5 atomes. Ce cycle se brise pour générer 2 cétones qui sont maintenant séparées. Le même mécanisme est obtenu avec HIO4. Comme il n’y a qu’un seul produit, le réactif est donc un cis-glycol cyclique.
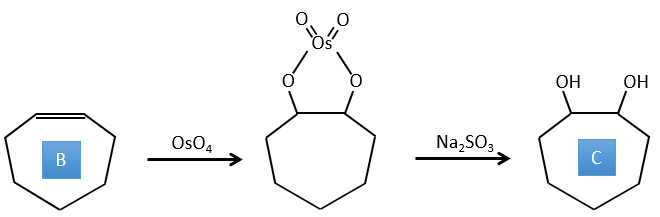
B→C: Le tétroxyde d’osmium est un réactif qui affecte spécifiquement C = C et qui génère un ester osmique, un peu comme le complexe formé par Pb(AcO)4 avec le cis-glycol. Na2SO3/H2O élimine l’osmium de la molécule pour obtenir le cis-diol. KMnO4 peut faire la même chose mais nous devons être dans des conditions basiques ou froides ou nous obtiendrons un diacide. Le réactif est donc un cycle de 7 carbone avec une double liaison entre deux des carbones.
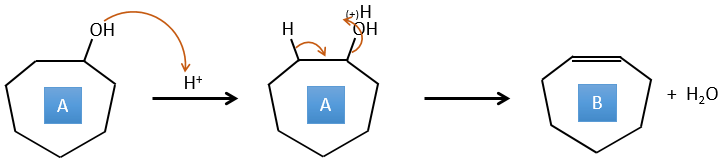
A→B: Cette réaction est simplement l’élimination d’une molécule d’eau du cycle qui donne une double liaison. On peut en déduire la différence de composition entre le réactif et le produit: C7H14O-C7H12=H2O.
A→G: C’est la réaction de chlorométhylation. Au cours de cette réaction, le formaldéhyde et l’acide chlorydrique forment un chlorométhanol stabilisé par ZnCl2. L’acide protonne l’alcool et l’anneau peut l’attaquer pour rejeter l’eau et lier le CH2Cl.
G→H: Un simple SN2 par CN– suivi de sa transformation en un acide carboxylique. Cette transformation se fait par des attaques successives de molécules d’eau sur la liaison carbone à l’azote.
H→I: SOCl2 est une molécule qui nous permet d’obtenir un chlorure d’acyle à partir d’un acide. Cela ne peut pas être fait avec HCl ou Cl2 car Cl– est un meilleur groupe partant que OH–. La réaction est suivie de la formation d’un amide primaire.
I→J: L’amide est réduit en une amine par LiAlH4. LiAlH4 peut générer H– qui attaque le carbonyle.
E+G→K: Une base prend le proton du bromophénol pour obtenir un nucléophile plus fort. IN SN2 a lieu entre les deux espèces pour les fusionner en une seule molécule.
G→M: La seconde étape de la réaction conduit à la formation de l’acide carboxylique comme c’était le cas dans la réaction G-> H. L’élément manquant sur M est le groupe nitro en méta. Cette position est favorisée en raison de l’effet de capteur mésomère du COOH à travers le CH2. L’effet est toutefois plus faible que pour un capteur mésomère directement en contact avec le cycle aromatique.
Chapitre 3 : Spectroscopie infrarouge
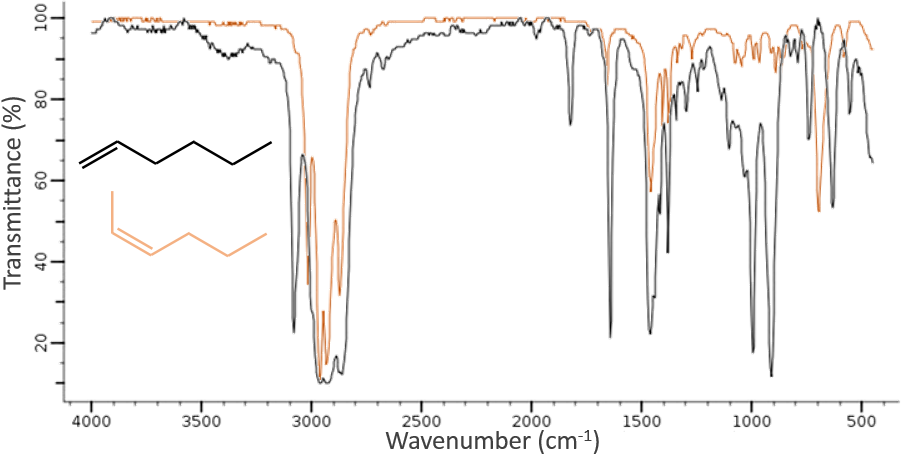
La spectroscopie infrarouge a un objectif différent de l’UV / visible. C’est un outil très puissant pour déterminer la structure des composés organiques. Un spectre IR est semblable à l’empreinte digitale d’une molécule et l’appariement des pics peuvent nous dire si une molécule est dans l’échantillon ou non. Le spectre montré ci-dessous est celui du 1-hexène en noir et du cis-2-hexène en orange. La différence de position de la double liaison conduit à une énorme différence dans le spectre IR.
La spectroscopie UV / visible implique des transitions électroniques tandis que la spectroscopie IR implique transitions entre états de vibration. Quant à l’UV / visible, on observe une série de bandes, et non des rayons, en raison des états de rotation subordonnés. Il existe deux principaux modes de vibration: l’allongement et la flexion.
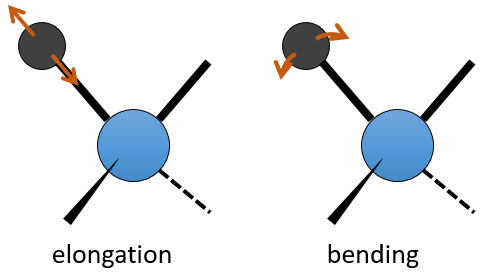
Une liaison entre deux atomes n’a pas une longueur constante. Les atomes vibrent autour de leur centre de masse avec une fréquence qui est caractéristique de la paire d’atomes. Il est le mode d’allongement: une vibration dans l’axe de la liaison. Cependant, les atomes sont souvent liés à plus d’un autre atome, en modifiant la fréquence de la vibration et donnant lieu à des processus d’allongement supplémentaires.
La flexion est une vibration hors de l’axe de la liaison. Elle conduit à une variation des angles entre les liaisons.
Pour une molécule de N atomes, le degré de liberté de la molécule est égale à la somme des degrés de liberté de chaque atome. Chaque atome a 3 degrés de liberté correspondant aux 3 coordonnées cartésiennes nécessaires pour décrire sa position dans la molécule. La molécule a donc 3n degrés de liberté. Parmi les 3n, 3 sont utilisés pour décrire les modes de translation et 3 sont utilisés pour décrire la rotation (2 si la molécule est linéaire). Il y a donc modes 3n-6 (ou 3n-5) modes de vibration pour chaque molécule. Par exemple, l’eau a 3 modes de vibration: 2 modes d’allongement et une mode de flexion.
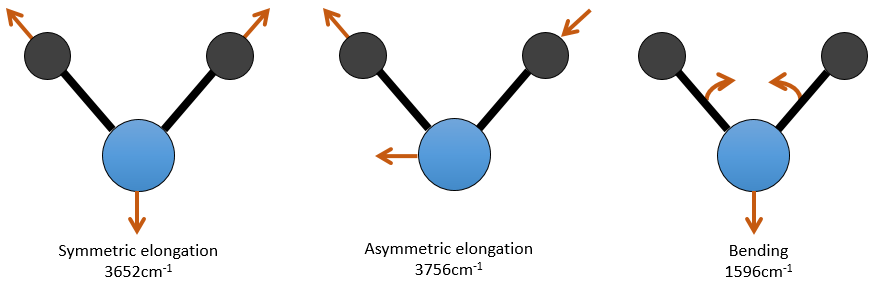
Un mode d’allongement est symétrique (le centre de la masse est décalé) et l’autre mode est asymétrique. Chaque mode représente une vibration spécifique, avec une longueur d’onde donnée. Les longueurs d’onde des modes d’allongement sont très proches les uns des autres et le mode de flexion a un nombre d’onde plus petit. Le fait que les modes d’élongation et les modes de flexion sont éloignés en termes de longueur d’onde et que les modes de flexion ont de plus petites longueurs d’onde sont des généralités que nous pouvons trouver pour toute liaison.
Il y a 3 atomes de carbone dans le dioxyde de carbone, mais 4 modes de vibrations comme le CO2 est linéaire (3n-5).
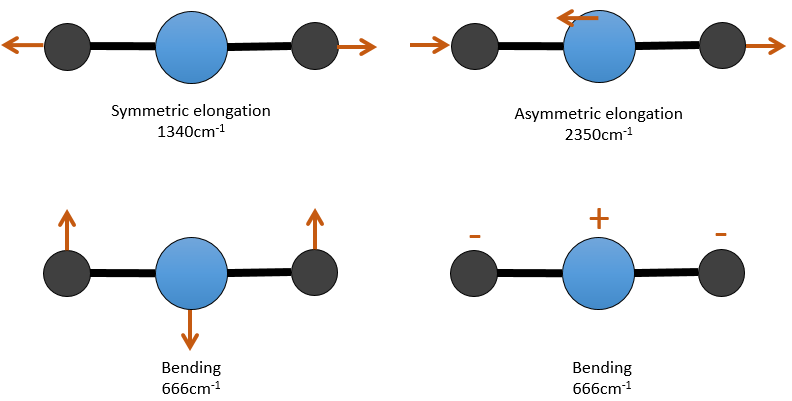
Il y a une forme d’allongement symétrique et une autre asymétrique et deux modes de flexion équivalentes (une dans le plan x-y et une dans la y-z). Les modes de flexion ont la même longueur d’onde à 666cm-1. Nous disons que ces modes de vibration sont dégénérées deux fois. En outre, l’allongement symétrique est inactive en IR parce que ce mode de vibration ne provoque aucune variation du moment dipolaire de la molécule.
Dans les grandes molécules, il est rare d’observer le nombre exact de modes de vibration (3n-6) parce que certaines modes viennent de la combinaison de deux ou plusieurs vibrations ou sont des harmoniques de puissants modes de vibration. Ces deux effets augmentent le nombre de bandes tandisque d’autres effets diminuent ce nombre par example:
– des bandes qui sont trop faibles pour être observés
– des bandes qui sont trop proche et qui fusionnent
– des bandes dégénérées
– des formes inactives de vibrations
– des modes avec des nombres d’ondes en dehors de la période analysée, généralement entre 4000 et 400 cm-1.
Il est possible de se rapprocher de la fréquence de vibration d’une liaison donnée avec la loi de Hooke qui considère la liaison comme un oscillateur harmonique simple entre deux masses M1 et M2 .
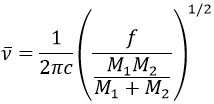
c est la vitesse de la lumière et f est une constante qui reflète la force de la liaison, dont la valeur est d’environ 5.105 dyne/cm (1 dyne =105N=105kg m s-2) pour les liaisons simples et deux et trois fois cette valeur pour les liaisons doubles et triples. f augmente de gauche à droite dans le tableau de Mendeleïev. Par exemple, la loi de Hooke donne une longueur d’onde de 3040cm-1 de la liaison C-H. Si on regarde les modes de vibration de CH2 à l’intérieur d’une chaîne carbonée, on trouve 6 modes de vibration (2 modes d’élongation et 4 modes de flexion).
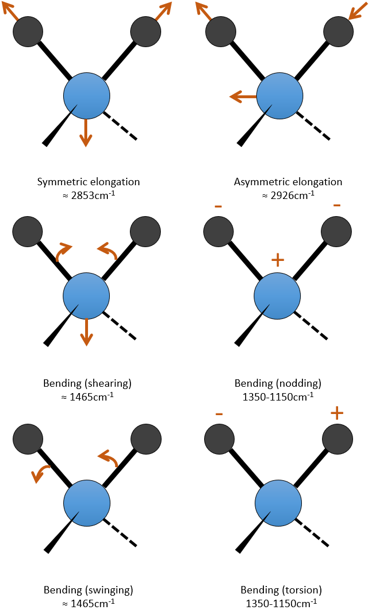
Rappelez-vous que la règle 3n-6 s’applique uniquement à des molécules complètes. Les modes d’allongement que nous observons ont des fréquences qui sont un peu inférieurs à ceux obtenus par la loi de Hooke (2926 et 2853cm-1 vs 3040cm-1) en raison de l’environnement de la liaison C-H qui ne sont pas pris en compte par Hooke.
De la loi de Hooke, il est évident que des atomes lourds vibrent à des fréquences plus petites. On peut à peine distinguer un spectre IR dans plusieurs régions en fonction de la paire d’atomes participant à la liaison et du type de liaison:
| 3800-2700cm-1: C-H, O-H, N-H | 1900-1500cm-1: C=C, C=O, C=N, N=O |
| 2300-2000cm-1: C≡C, C≡N | 1300-800cm-1: C-C, C-O, C-N |
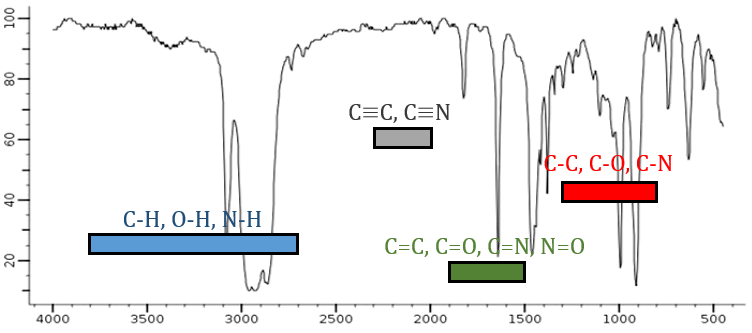
Sur la figure, nous pouvons donc en déduire déjà qu’il n’y a pas de liaison triple (région grise) dans notre molécule (le 1-hexène à partir de début), qu’il y a des liaisons doubles (région verte) et des liaisons simples (régions de bleu et rouge) . Cependant, nous ne pouvons pas encore déterminer quel pic correspond à laquelle les vibrations.
Interactions couplées
Lorsque deux oscillateurs partagent un atome commun, ils agissent rarement comme un des oscillateurs simples sauf si leurs modes de vibrations sont très différents. Le couplage entre deux modes de vibration produit deux nouveaux modes de vibration à des fréquences de plus en plus petite que celle en absence d’interaction.
Par exemple, dans le CO2, l’allongement asymétrique (2350cm-1) et l’allongement symétrique (1340cm-1) sont le résultat d’un couplage entre les deux oscillations C = O, l’ une raccourcit lorsque l’autre une allonge.
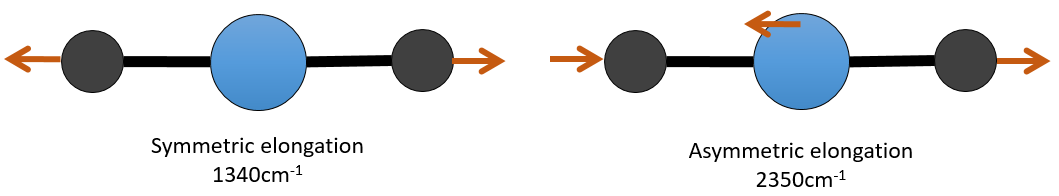
De ce que nous avons vu précédemment, un C = O devrait vibrer entre 1900-1500 cm-1, mais ce n’est clairement pas le cas ici. Le couplage entre les deux vibrations C = O a pour effet de déplacer la vibration vers les grandes fréquences (seul l’asymétrique est visible). Le couplage devient négligeable quand un ou plusieurs atomes de carbone séparent les agents de liaison. Deux carbonyles séparés par un ou plusieurs atomes de carbone montrerait une absorption vers 1725cm-1.
par conséquant pour avoir un couplage il faudrait que les vibrations partagent un atome et qu’elles aient des fréquences similaires. Cependant des interactions sont possibles entre les vibrations fondamentales et les vibrations harmoniques et/ou les vibrations de combinaison. Un tel couplage est appelé une résonance de Fermi. En fin pour le CO2, l’allongement symétrique n’est pas activé dans IR mais qui peut être observé dans le spectre Raman à 1340cm-1. En fait, il existe deux bandes à 1286cm-1 et 1388cm-1 en raison du couplage de la vibration avec le harmonique des modes de flexion (666cm-1). La première harmonique est donc à 1332cm-1 et interagit avec le mode d’allongement symétrique à 1340cm-1.
Des liaisons hydrogène
Les liaisons hydrogène diminuent les fréquences des liaisons et généralement élargit et augmente leurs bandes. L’importance de cet effet dépend de la force de la liaison H. Une liaison H est solide quand elle est dans la direction exacte de la paire libre de l’atome électronégatif. La force de la liaison dépend également de la distance entre les atomes, de l’atome et le fait que si un cycle peut être formé par la liaison H.
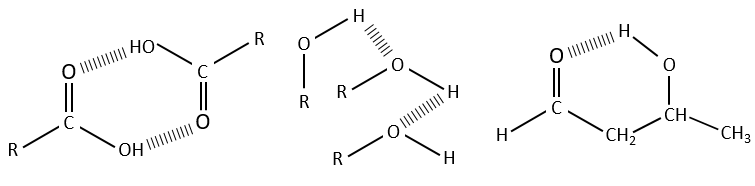
La diminution de la fréquence varie de 300 à plus de 500 cm-1 si la liaison H est intermoléculaire et est de moins de 100 cm-1 à plus de 300cm-1, si la liaison H est intramoléculaire. Les liaisons H sont souvent présentes entre une molécule et le solvant. Il est donc important d’indiquer le solvant et la concentration du composé sur un spectre. La présence d’eau dans l’échantillon a un effet visible sur les spectres.
Analyse d’un spectre :
Un spectre IR montre la transmittance en fonction de la longueur d’onde/nombre d’ondes. Le spectre est à lire de haut en bas avec des bande descendant bas dans le spectre. Une analyse précise d’un spectre IR n’est pas concevable. Les couplages, l’absence de modes de vibration et de la largeur de certaines bandes,rendent la détermination de la nature exacte des bandes doifficile. Cependant, comme il a été dit précédemment, un spectre IR est comme l’empreinte digitale d’un composé et si le spectre se superpose avec le spectre d’une molécule connue (avec le même solvant, la même concentration et la même configuration), le travail est fait. Dans d’autres situations, un spectre IR est utilisé en combinaison avec MS, UV et RMN. Le spectre, habituellement entre 4000 et 400 cm-1 peut être divisé en deux régions importantes pour analyser: une entre 4000 et 1300 cm-1 , appelées zone des groupes fonctionnels, et une en desous 1900 cm-1 qui montre les modes de flexion.
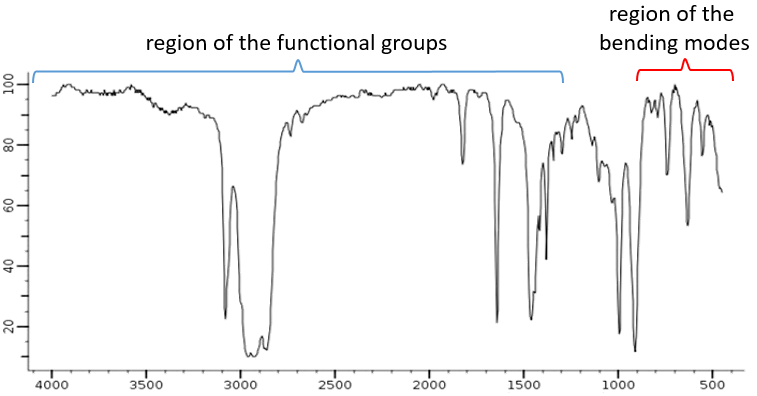
Les modes d’allongement de groupes fonctionnels avec OH, NH, C = O se trouvent dans la zone des groupes fonctionnels. Si cette région est vide, nous pouvons supposer que la molécule ne porte pas l’un d’eux. Dans de rares cas, une bande faible et/ou très large peut être confondue avec une absence de bande. Et encore il faut faire attention aux bandes de faible intensité qui peuvent aussi être des harmoniques ou des bandes de combinaison.
Les absorbances caractéristiques :
Il serait trop long de décrire toutes les bandes qui peuvent être observés sur un spectre IR. Le but ici est de montrer les bandes très caractéristiques qui peuvent être facilement repérés sur le spectre et d’avoir une idée générale de ce qui est dans la molécule et ce qui n’est pas. En combinaison avec un spectre de masse, il sera possible de déterminer la structure correcte de la molécule analysée.
Vibrations de hautes fréquences :
La région du spectre ci-dessus 2850 cm-1 est riche en informations qui peuvent facilement être étudié. Dans cette région, nous trouvons les bandes d’allongement de Y-H, Y = C, O, N. Les bandes sont généralement intenses et ne sont pas cachées, même si une autre bande est à proximité. La position d’une bande dépend de l’environnement direct de la liaison. Lorsque cela est nécessaire, nous écrivons cette liaison (simple, double ou triple) pour démarquer = C-H, à savoir une liaison hydrogène à un carbone avec une double liaison, du -C-H par exemple, à savoir une liaison hydrogène à un carbone avec une liaison simple.
Une première indication sur la nature du composé se trouve dans le voisinage de 3000 cm-1 où les liaisons C-H absorbent. La différence de fréquence entre aromatique = C-H et -C-H aliphatiques est petite mais est caractéristique:
Les C-H méthylique se trouvent en 2 bandes à 2962 cm-1 et 2872 cm-1 et les C-H méthylénique se trouvent un peu plus bas (2926 et 2853 cm-1). Les = C-H se trouvent légèrement au-dessus 3000 cm-1. Il est souvent synonyme d’un aromatique, mais il peut aussi être simplement un alcène (ou doubles liaisons conjuguées). Sur le spectre de l’hexène, le pic à 3080 cm-1 est caractéristique de la double liaison et les pics pour le C-H sont cachés dans le grand groupe. Trois petits pics sont toutefois visibles autour des valeurs citées ci-dessus pour des -C-H méthylique méthylénique.
Il est donc facile de deviner la présence d’un cycle aromatique à partir des bandes -C-H mais il n’est pas facile de déterminer si la bande d’absorbance = C-H provient d’un cycle aromatique ou d’un alcène: car aussi bien le = C-H alcène que le =C-H aromatique montrent une bande intense dans la région des basses fréquences, respectivement entre 1000-650 cm-1 et 900 à 675 cm-1. Les composés aromatiques (et hétéro aromatiques) vont également montrer 3 pics autour de 1600 cm-1, 1500-1400 cm-1 et 1300-1000 cm-1(à noter que les alcènes conjugués montrent également ce genre de bandes). Toujours dans le spectre de l’hexène, le pic entre 1300-1000 cm-1 n’est pas présent et le pic de flexion est inférieure à 675 cm-1.
Les liaisons C-H des aldéhydes vibrent à des petits nombres d’onde, entre 2830 et 2695 cm-1 parce que le carbonyle prend les électrons de la C-H et que cette liaison devient plus long, ce qui fait qu’elle vibre plus lentement. Le pic n’est pas très intense et peut être doublée en raison d’une résonance de Fermi avec la première harmonique du mode de flexion à 1390 cm-1.
Les liaison ≡C-H absorbent à des fréquences supérieures à 3100 cm-1, entre 3333 cm-1 et 3267 cm-1 mais ne sont pas les seules espèces absorbantes au dessus de 3100 cm-1. Nous pouvons aussi trouver -O-H et les bandes d’élongation N-H ainsi que des liaisons hydrogène. Les bandes de ≡CH sont généralement plus minces que les bandes pour -O-H et -N-H et sont intenses.
Les Bandes d’allongement -O-H
Les bandes -O-H sont grandes et intense, et très grand dans le cas des acides carboxyliques. En effet, ces grandes bandes sont le résultat des liaisons H et la formation de dimères, lorsque la concentration d’alcool / acide sont bien dilué.
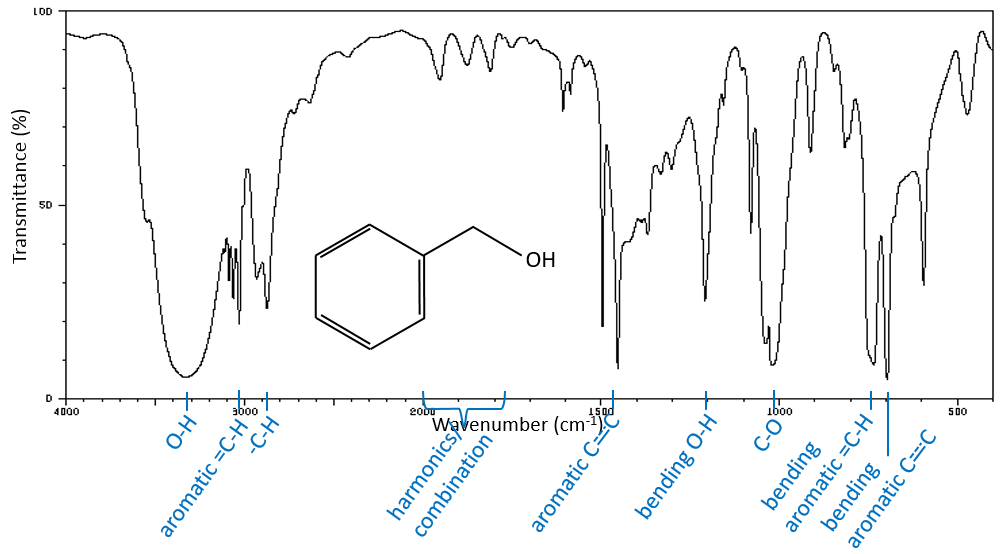
Les -O-H libres donnent des bandes petites et minces comprises entre 3650 et 3550cm-1 pour les alcools et autour de 3550cm-1 pour les acides, mais sont facilement effacées par les larges bandes de H: O-H qui se répandent sur des centaines de cm-1 sont de façon plus intense. Ces grandes bandes doivent être centrées entre 3550 et 3200 cm-1 (pour l’alcool benzylique, il est à 3320 cm-1) pour les alcools et 3000 cm-1pour les acides.
Les bandes d’allongement -N-H
Les bandes de fréquences élevées pour les amines aliphatiques sont assez faibles entre 3400 et 3250 cm-1. Il y a deux bandes dans le cas des amines primaires et une bande dans le cas d’une amine secondaire. Les modes d’élongation N-H sont légèrement déplacées vers les grandes fréquences si l’amine est aromatique. Le spectre est différente dans le cas des sels d’amine. Les ions ammonium donnent une bande d’absorption grande et intense entre 3300 et 3030 cm-1. La fréquence diminue avec le degré de l’amine: amine primaire: 3000-2800 cm-1, amine secondaire: 3000-2700 cm-1 et amine tertiaire: 2700-2250 cm-1).
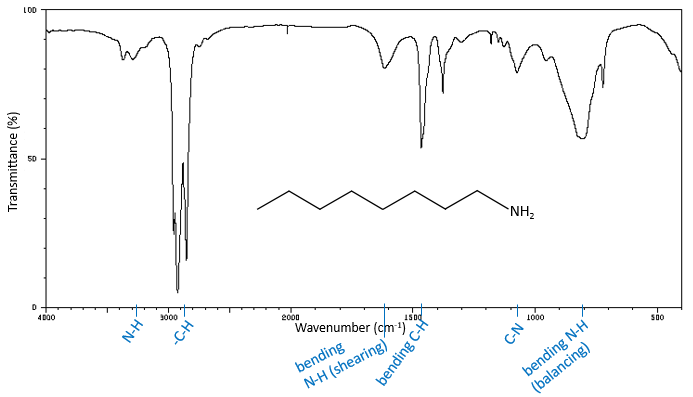
En l’absence de liaison H, l’amide primaire présente deux bandes autour de 3520 et 3400 cm-1, tandis que les amides secondaires montrent une bande d’absorbance entre 3500 et 3400 cm-1. En plus dans les solutions ou dans les solides concentrés, les liaisons H déplacent les bandes vers les petites fréquences, respectivement autour de 3350 et 3180 cm-1 pour les amides primaires et plusieurs bandes entre 3330 et 3060 cm-1 pour les amides secondaires. La présence de plusieurs bandes au lieu d’une seule bande est expliqué par la formation d’un variateur de lumière d’amides.
fréquences de milieu de gamme :
Entre 2850 et 900 cm-1, nous pouvons trouver des bandes caractéristiques des vibrations d’allongement entre deux atomes autres que H et quelques vibrations de flexion de Y-H qui peuvent confirmer les observations de la région des hautes fréquences du spectre IR. Nous allons nous concentrer sur les bandes d’élongation. Les liaisons S-H produisent une petite bande d’absorption entre 2600 et 2550 cm-1. Cette bande est solitaire dans cette région du spectre, mais dans des solutions diluées de la bande peut être trop petite pour être détectée. C≡C montre une petite bande d’absorbance entre 2260 et 2100 cm-1. Cette région est dédiée aux liaisons triples (C≡N montre une bande entre 2260 et 2240 cm-1), mais les harmoniques et les bandes de combinaison peuvent être trouvées dans cette région aussi. Les bandes de C = C sont ou d’intensité moyenne ou faible entre 1667 et 1640 cm-1 pour alcènes qui ne sont pas conjugués. Ces alcènes se trouvent entre 1650 et 1600 cm-1. Les alcènes cumulés (c = c = c) absorbent entre 2000 et 1900 cm-1.
vibrations Allongement de C=O
L’absence d’absorbance entre 1870 et 1540 cm-1 est synonyme de l’absence de carbonyle. Dans cette région, la position du pic dépend de la conjugaison du groupe carbonyle et des substituants (qui substituant et sa masse) et les liaisons possibles H. Une cétone aliphatique se trouve à 1715 cm-1. Les modifications de l’environnement du cétone peut induire soit une augmentation d’une diminution du nombre d’onde du pic qui dépend de la prédominance de l’induction ou de l’effet de résonance. L’effet inductif réduit la longueur de la liaison C = O, ce qui conduit à l’augmentation de sa fréquence de vibration. Une résonance augmente la longueur de la liaison C = O, entraînant le déplacement du pic vers le sommet C-O qui est à une fréquence plus basse. Par exemple, Cl a un effet inductif prédominant et le pic de la C = O est à 1815-1785 cm-1. Le groupe carbonyle d’un amide primaire vibre à 1695-1650 cm-1 parce que l’effet de résonance est prédominante. Il y a deux bandes pour les amides primaires et une seule pour les autres. Carbonyles conjugués avec des alcènes ont des pics d’absorption à plus petits nombres d’onde que la cétone aliphatique entre 1685 et 1666 cm-1. Un aldéhyde absorbe un peu plus élevé que la cétone (1740-1720 cm-1). Un acide absorbe fortement (de façon plus intense qu’une cétone) autour de 1760 cm-1, mais des liaisons H dans les gradateurs d’acides peut déplacer le pic vers les petites fréquences (1720-1706 cm-1). les esters absorbent à 1750-1735 cm-1. Un halogénure d’acyle présente une forte absorption entre 1815 et 1785 cm-1.
Les liaisons H diminuent la fréquence de vibration des carbonyles. L’effet est faible pour les obligations intermoléculaires H (une dizaine de cm-1), mais peut être important pour les obligations H intramoléculaires.
vibrations Allongement de C-O
Dans le spectre du milieu de gamme, entre 1300 et 900 cm-1 du spectre est généralement compliqué. Il est le « impression numérique » du composé où nous pouvons trouver les bandes d’alcools C-C-O qui devrait être accompagné de bandes O-H à grandes fréquences.
La liaison C-O des alcools présente une bande intense à cause des vibrations de l’allongement entre 1260 et 1000 cm-1. Ce mode de vibration est généralement couplée à la prochaine carbone, à titre d’élongation C-C-O asymétrique. Le COC des éthers et des systèmes d’ époxydes peuvent également être trouvés dans cette région du spectre, entre 1150 et 1086 cm-1 (pour son allongement asymétrique, symétrique étant très faible, car il ne modifie pas le moment dipolaire) comme une bande intense ou plusieurs bandes si les carbones à proximité sont ramifiées. Si un côté est un groupe aryle, on observe une bande intense (allongement asymétrique) à 1275-1200 cm-1 et une bande intense (allongement symétrique) à 1075-1020 cm-1). Enfin, les éthers de vinyle présentent des groupes similaires (1225-1200 cm-1 et 1075-1020 cm-1).
Les acides donnent deux bandes d’intensités inégales dans le spectre , en plus de celui de C = O provenant d’une interaction entre le mode d’allongement de C-O et le mode de flexion d’O-H. Nous avons des bandes de C-O-H. La bande la plus intense entre 1315 et 1280 cm-1 est appelée la bande d’élongation C-O. La deuxième bande, entre 1440 et 1395 cm-1 est le groupe appelé la bande de flexion O-H. Cette bande tombe dans la même région que le mode de CH2 de cisaillement. Le carboxylate donne également deux bandes d’intensités inégales, la plus intense une entre 1650 et 1550 cm-1 et l’autre autour de 1400 cm-1.
Une particularité des esters est leur bande intense d’absorption à la place du mode de fonctionnement des cétones d’allongement. Cette bande n’est pas le groupe C = O, qui est déplacée vers les grandes fréquences mais l’une des deux bandes de C-O. Le groupe C = O est en effet entre 1750 et 1735 cm-1 et 20 cm-1 inférieur si le groupe carbonyle est conjugué. La vibration C-O est couplé avec les deux côtés de la liaison, ce qui conduit à deux bandes d’absorption. Le plus intense est une C-C (= O) -O donnant un pic entre 1300 et 1000 cm-1. Les esters d’acides aromatiques absorbent fortement entre 1310 et 1250 cm-1. L’autre bande entre 1111 et 1031 cm-1 provient du couplage O-C-C. Sa fréquence dépend du substituant de l’ester. Esters d’alcools primaires ont les fréquences les plus basses (1064-1031 cm-1 ), tandis que les esters d’alcools secondaires et d’aromatiques absorbent autour de 1100 cm-1 et 1111 cm-1 respectivement.
vibrations Allongement de NO :
les groupes nitro ont un symétrique et un mode asymétrique d’élongation de la vibration. Le mode asymétrique donne une bande intense entre 1661 et 1499 cm-1. Le mode symétrique donne une bande comprise entre 1389 et 1259 cm-1. Les nombres d’ondes des deux bandes dépendent du substituant du groupe nitro. Dans le cas d’un alcane, les bandes doivent être observées autour de 1550 et 1372 cm-1, respectivement. Un groupe électronégatif sur le carbone alpha augmente la fréquence tandis qu’une conjugaison diminue la fréquence.
Les nitrates montrent 2 bandes intenses d’allongement pour N = O (une asymétrique et une symétrique) entre 1300-1255 cm-1 et 1660-1625 cm-1. Les nitrites ont deux bandes ainsi, l’un pour les cis (1625-1610 cm-1) et un pour l’isomère trans (1680-1650 cm-1). Composés nitroso montrent une bande d’absorption entre 1585 et 1539 cm-1.
Modes d’ allongement de C = S :Les modes d’allongement de C-S sont présents dans les basses fréquences et C = S légèrement au-dessus de cette région, entre 1250 et 1020 cm-1 . Cette bande est moins intense que celle d’un groupe C = O, car il est moins polaire.
Modes d’ allongement de S = O :
Les liaisons S = O produisent des bandes intenses en général. La liaison S = O de sulfoxydes vibre entre 1070 et 1030cm-1. Sulfones montrent deux bandes d’absorption entre 1350-1300cm-1 et 1160-1120cm-1. Les bandes sont décalées vers des fréquences plus importantes dans le cas d’un chlorure de sulfonyle (1410-1380cm-1 et 1204-1177cm-1) ou d’un sulfonamide (1370-1335cm-1 et 1170-1155cm-1).
Pliage dans la région de milieu de gamme
Plusieurs liaisons avec les modes d’allongement de la haute fréquence, typiquement Y-H, les liaisons ont modes de flexion qui tombent dans la même région que d’autres modes d’allongement (Y-Z). Ils compliquent la lecture du spectre, mais peuvent également confirmer des pics de la région des hautes fréquences.
Modes de flexion de C-H
Les modes de flexion des liaisons méthyléniques C-H ont été vu au début de ce chapitre. Un mode est dans la région basse des fréquences 720cm-1 et les trois autres sont dans la région de milieu de gamme. Le mode de cisaillement a une wavenumber presque fixe de 1465cm-1, mais les deux derniers modes (nod et torsion) donnent des pics entre 1350 et 1150cm-1. Dans un cycle, ces fréquences sont légèrement diminué. Méthyliques C-H ont deux modes de flexion: l’une où les trois H oscillent en phase et un dans lequel un H ne sont pas en phase. Le mode symétrique (en phase) donne un pic à 1375cm-1 et le mode asymétrique à 1450cm-1, à peu près à la même fréquence que le mode d’un groupe C-H méthylénique cisaillement. = C-H 1415cm à absorber alcènes-1 en raison d’un mode de cisaillement. Des hydrocarbures aromatiques ont un mode de flexion hors du plan qui donnent des pics intenses dans la région des basses fréquences et un mode de flexion dans le plan donnent des pics entre 1300 et 1000cm-1.
Pliage O-H
Pics d’alcools ne sont pas très caractéristique et se trouvent dans la région comprise entre 1420 et 1330cm-1. S’il y a un proton sur l’atome de carbone portant le groupe -OH (par exemple les alcools primaires et secondaires), il existe un couplage qui donne deux bandes autour 1420 et 1330cm-1. Les alcools tertiaires donnent un seul pic. Dans le cas des acides, il y a une interaction entre le mode d’allongement de C-O et le mode de flexion de l’O-H (décrit dans la section C-O modes d’allongement). Le mode de flexion C-O-H devrait être trouvé entre 1440cm-1 et 1395cm-1.
Pliage N-H
Les amines primaires ont un mode de flexion qui donne un pic de moyenne à haute intensité entre 1650cm-1 et 1580 cm-1. Le mode de flexion des amines secondaires est difficilement détectable à l’exception des amines aromatiques secondaires qui absorbent environ 1515cm-1. Dans le cas des amides, le mode de flexion N-H doit être observée par une bande comprise entre 1655 et 1590cm-1 avec une intensité façon plus petite que l’intensité du pic C = O. Comme C = O absorbe dans la même région, les pics de l’allongement C = O et de flexion N-H peuvent fusionner.
Les basses fréquences région
Dans cette région, on trouve principalement les modes de flexion, mais certains modes d’élongation sont également présents. À mon avis, cette région est moins intéressant que les régions de fréquences hautes et moyennes et est assez difficile à lire parce que plusieurs modes ont de grandes gammes de fréquences possibles qui se chevauchent avec d’autres modes. Les plupart des pics caractéristiques sont les pics pour les gradateurs et pour les aromatiques. Entre 900 et 650 cm-1, une bande large et intense caractéristique d’un variateur de lumière d’acide, un amide ou une amine. Dans la même région, l’absence de pics intenses indique que la molécule ne soit pas un groupe aromatique.
modes de flexion du CH
Le pic du mode de méthylénique -C-H balancer à 720cm-1 a une faible intensité ou non visible. Une bande très intense (généralement les plus intenses du spectre) est visible entre 1000 et 650 cm-1 en cas d’alcène est présent dans la molécule, provenant d’un mode de flexion hors du plan. Il est habituellement ce genre de flexion hors du plan qui donne des bandes intenses dans la région des basses fréquences. Un pic très intense peut donc être observée pour les aromatiques entre 900 et 650 cm-1. Les alcynes donnent un pic épais et intense à des fréquences plus basses, entre 700 et 610 cm-1.
D’autres modes de flexion
Le mode de flexion des alcools donne un pic d’épaisseur entre 769cm-1 et 650 cm-1 en plus des modes de flexion de la région des fréquences moyennes. Dimères d’acides donnent une très large bande (pas aussi large que la bande de hautes fréquences qui donnent les acides, mais il est caractéristique) d’intensité moyenne centrée près 920cm-1. Amides donnent également une épaisse bande d’intensité moyenne, mais entre 800 et 666cm-1, encore une fois en raison d’une flexion hors du plan. Cette courbure est plus forte et à des fréquences plus élevées pour les amines (909-666cm-1).
La flexion hors du plan des amines et des amides de générer une moyenne / bande intense respectivement entre 1 et 909-666cm 800-666cm-1. les groupes nitro semblent montrer un mode de flexion entre 763 et 690cm-1.
modes de Allongement de la région basse des fréquences
Une bande d’allongement de la N-O apparaît entre 870-833cm-1 à partir des groupes nitro, et entre 850-750cm-1 à partir nitrites. C-S absorbe entre 700 et 600 cm-1.
Enfin, les halogènes ont leurs modes de allongements dans les basses fréquences. Dans une chaîne aliphatique, C-Cl absorbe entre 850 et 550 cm-1, C-Br entre 690 et 515cm-1, C-I entre 600 et 500 cm-1 et C-F entre 1,400 et 730cm-1. La bande de C-F est forte et grande. Si le substituant est un groupe aromatique, les bandes sont déplacées vers les grandes fréquences, au-dessus 1000cm-1 dans le cas des chlorobenzènes.
Chapitre 2: Méthodes spectroscopiques
Les méthodes spectroscopiques sont destinées à déterminer la composition des échantillons contenant une ou plusieurs espèces sur la base de leurs niveaux d’énergie. Nous avons vu que les électrons des atomes n’ont pas accès à toutes les valeurs de l’énergie. Ils sont limités à quelques valeurs de l’énergie, l’énergie’S (niveaux orbitales 1s, 2s, 2p, etc), sur lesquels ils sont distribués suivant la règle de Fermi en commençant par les niveaux d’énergie avec des énergies plus faibles et en remplissant les niveaux jusqu’à ce qu’il n y ai plus d’électrons à placer.
Les électrons de plus haute énergie peuvent être excités et atteindre les orbitales d’énergies plus élevées si elles reçoivent le montant exact des AE de l’énergie séparant les deux niveaux d’énergie, sous forme de chaleur ou de photons. Nous disons que l’électron passe de son état fondamental à un état excité.
Quand un électron excité revient à son état fondamental il génère un photon avec la même énergie AE = hv. La fréquence du photon est caractéristique de l’atome ou de la molécule à partir de laquelle il est émis. La ν de fréquences, comme une unité de mesure, est le nombre de fois que le processus se produit sur une période d’une seconde. Dans le cas des photons c’est le nombre d’ondes qui passe par un point durant une seconde (unités : Hertz). La longueur d’onde λ = c/ν est la longueur d’une onde ou la distance entre deux sommets, en mètres. Le nombre d’onde n est égal à l’inverse de la longueur d’onde (1/λ, les unités : cm-1). La différence entre la fréquence et le nombre d’onde est que le nombre d’onde n’a rien à voir avec la vitesse de l’onde. Il est donc utile pour des situations où la vitesse ou la fréquence ne sont pas fixes.
Par exemple lorsque nous chauffons un métal il change de couleur parce que les électrons excités émettent des photons dans la gamme du visible. Le spectre du visible est composé de toutes les couleurs que les humains peuvent voir. Chaque couleur correspond à un photon d’une longueur d’onde donnée. Le violet est la couleur visible avec la plus petite longueur d’onde (390 nm) ou le plus grand nombre d’ondes et le rouge est la couleur visible qui possède la plus grande longueur d’onde (780 nm) ou le plus petit nombre d’onde. Ironiquement la couleur que nous voyons sur un matériau donné n’est pas la couleur que le matériau absorbe mais plutôt les couleurs qui n’ont pas été absorbées et qui sont donc réfléchies par le matériau vers notre œil.
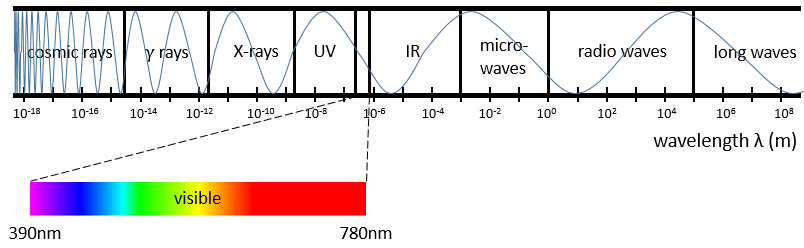
Les radiations ne sont pas limitées au spectre visible. Il existe des photons de longueur d’onde au-dessus de 780 nm et en-dessous de 390 nm. Les radiations avec un nombre d’onde plus petite (la longueur d’onde plus grande) que le spectre visible sont appelées infrarouge (le rouge étant la couleur avec le plus petit nombre d’onde = une avec la plus grande longueur d’onde). Si nous continuons de diminuer le nombre d’onde (ou d’augmenter la longueur d’onde) nous atteignons les micro-ondes, puis les ondes radio. De l’autre côté du spectre visible l’on retrouve les rayonnements ultraviolets (le violet le plus grand nombre d’onde ayant le spectre visible), puis les rayons X et les rayons gamma enfin. Comme l’énergie d’un photon est E = hv les photons gagnent de l’énergie quand nous allons à partir des ondes radio vers les rayons gamma. Les rayons X ont assez d’énergie pour extraire un électron profond (pas d’électron de valence) à partir d’un atome. Pas besoin de dire les dommages que les rayons gammas, à partir d’éléments radioactifs, peuvent faire.
Les transitions électroniques dont nous avons parlé précédemment correspondent à des radiations dans l’UV/spectre visible. Ces transitions ne sont pas les seules transitions qui peuvent avoir lieu. Entre chaque niveau électronique nous trouvons les niveaux de vibration et entre les niveaux de vibrations, nous trouvons les niveaux de rotation. Les transitions entre les niveaux de vibration sont de l’ordre de l’IR tandis que les transitions du niveau de rotations sont dans la gamme des micro-ondes. Ces transitions conduisent à une émission de chaleur et c’est exactement comme ça que le four à micro-ondes fonctionne: la nourriture est bombardée par des radiations dans la gamme des micro-ondes et la rotation des molécules de la nourriture génère le chauffage de celle-ci.
La spectroscopie
L’absorption des radiations d’une solution dépend donc de sa composition. Nous pouvons mesurer si une solution absorbe un rayonnement donné ou comment cette solution émet des photons lorsque ses molécules sont excitées.
La spectroscopie d’absorption
L’absorbance A d’une solution est sa capacité à bloquer/absorber les radiations. L’absorbance A dépend de la longueur d’onde(λ) du rayonnement.
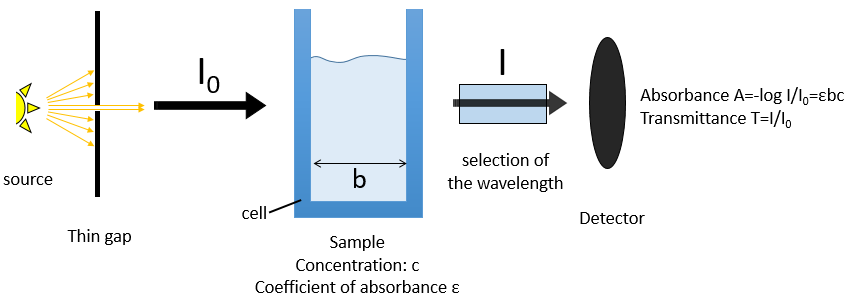
Si l’on considère un rayon entrant avec une intensité I0 qui se dirige vers une cellule de longueur b contenant une solution avec une concentration c, l’intensité I du rayonnement derrière la cellule est réduite par dI proportionnellement à la longueur de la cellule (une cellule plus longue absorbe plus) et de la concentration de la solution (une solution plus concentrée absorbe plus).
![]()
E est le coefficient d’absorbance de la solution qui ne dépend pas de la concentration ni sur la taille de la cellule et dc est la concentration de la solution à travers laquelle le rayon passe, à savoir la différence de concentration entre l’intérieur et l’extérieur de la cellule. Nous pouvons réécrire cette expression comme :
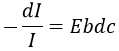
et on peut intégrer dI et dc entre l’émission du rayon et sa détection, soit à partir de I = I0 à I = I et de c = 0 à c. Il donne :
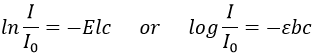
L’absorbance est donc donnée par la loi de Beer-Lambert
![]()
On peut aussi définir la transmittance qui est T = I/I0.
L’absorbance est donc directement proportionnelle à la concentration de la solution. Des déviations de la ligne droite peuvent être observées pour les solutions concentrées (c≠activité) ou si certaines molécules de l’échantillon sont impliquées dans un équilibre.
Les parois de la cellule sont fabriquées de telle manière à laisser passer un maximum de rayons. Ils sont généralement en quartz (transparent entre 200 et 380 nm). Une variation de l’intensité du rayon peut cependant être observée mais peut être déterminée en utilisant une solution blanche, à savoir une solution contenant seulement le solvant, avec c = 0 ou une référence. Le détecteur peut être configuré pour définir T = 1 (ou A = 0) avec la solution « blanc ». Comme l’absorbance est directement proportionnelle à la concentration de la solution on peut déterminer la concentration de l’échantillon si nous avons mesuré l’absorbance de notre solution blanc et d’une solution étalon dont la concentration est connue.
Si plusieurs espèces se trouvent dans la solution l’absorbance est la somme des absorbances individuelles.
![]()
Pour déterminer les concentrations nous avons besoin de répéter l’expérience avec plusieurs longueurs d’onde. Ne pas oublier que l’epsilon dépend de la longueur d’onde.
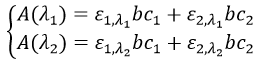
Nous pouvons ainsi déterminer C1 et C2 si les εi sont connus.
Installation expérimentale :
L’instrument est composé de 5 grandes parties :
– une source stable de l’énergie rayonnante.
– un dispositif transparent destiné à contenir l’échantillon.
– un système pour sélectionner une gamme de spectre.
– un détecteur qui peut transformer le signal lumineux en un signal électrique.
– un dispositif pour traiter le signal.
La source du rayon doit être une lampe monochromatique envoyant uniquement des photons de longueur d’onde donnée. Il peut être par exemple une lampe à cathode creuse ou une source laser. Cependant il y a toujours une petite gamme de longueurs d’ondes qui est émise. L’impact peut être limité si la longueur d’onde est de sorte que les coefficients de ε ne varient pas trop sur cette gamme de longueurs d’onde.
La cellule est généralement en quartz et sa taille est généralement de 1 cm de long pour la spectroscopie des solutions. Pour les gaz l’épaisseur est comprise entre 1 et 100 mm mais le rayon peut passer plusieurs fois dans la cellule, avec l’utilisation de miroirs, donnant chemins optiques jusqu’à 120m. Il est également possible de permettre un écoulement de gaz.
La sélection de la longueur d’onde est réalisée par une combinaison de prismes/miroirs et les lacunes (gaps) minces. Les prismes séparent les longueurs d’onde de la lumière incidente et les lacunes (gaps) permettent seulement une petite gamme de longueurs d’onde pour passer vers le détecteur.
Le détecteur peut également introduire des erreurs dans la mesure. L’intensité relative de ces erreurs est grande lorsque A →0 ou lorsque A est grand. Encore une fois, un bon choix de longueur d’onde limite les erreurs expérimentales.
Spectroscopie moléculaire :
Une autre façon d’utiliser les propriétés d’absorption des solutions consiste à balayer une gamme de longueurs d’onde et à détecter l’absorbance. Il nécessite une source de lumière capable de changer de longueur d’onde. S’il n’y avait que les niveaux électroniques nous obtiendrions des rayons discrets correspondants à la quantité exacte d’énergie de la transition entre l’état fondamental de l’électron et un état excité. Cependant les sous-niveaux de rotation et de vibration transforment les rayons en bandes. Les bandes sont amincies si nous effectuons la spectroscopie des gaz. Dans le cas de la spectroscopie d’absorption UV-visible, cette technique est surtout utilisée quantitativement. Par exemple nous pouvons suivre l’évolution d’une réaction à travers le spectre d’absorbance. Si les produits et les réactifs absorbent à différentes longueurs d’onde, l’intensité des pics caractéristiques des produits augmente avec le temps tandis que les sommets des réactifs diminuent en taille.
Chromophore : c’est un groupe organique insaturé covalent qui absorbe dans l’UV (ex: C = C, C = O, NO2) avec des électrons qui peuvent être délocalisés. Les photons excitent les électrons qui sautent entre les niveaux d’énergie qui sont des orbitales π étendues, créés par une série d’alternance de liaisons simples et doubles, souvent dans des systèmes aromatiques.
Auxochrome : un groupe saturé avec des électrons libres qui modifient l’intensité et la longueur d’onde de l’absorption lorsqu’il est lié à un chromophore.
Déplacement bathochromique : déplacement de l’absorption vers les longueurs d’onde plus grande (déplacement vers le rouge) en raison d’un effet de solvant ou d’une substitution.
Déplacement hypsochromique : déplacement de l’absorption vers les longueurs d’onde plus petites (déplacement vers le bleu) en raison d’un effet de solvant ou d’une substitution.
Hyperchromicité : augmentation de l’intensité de l’absorption.
Hypochromicité : diminution de l’intensité de l’absorption.
Chapitre1: Réactions de substitution sur les cycles aromatiques
Les cycles aromatiques, tels que le benzène, sont très stables en raison de leur énergie de résonance. Par conséquent, il est très difficile d’ « ouvrir » le cycle habituel par une réaction d’addition.
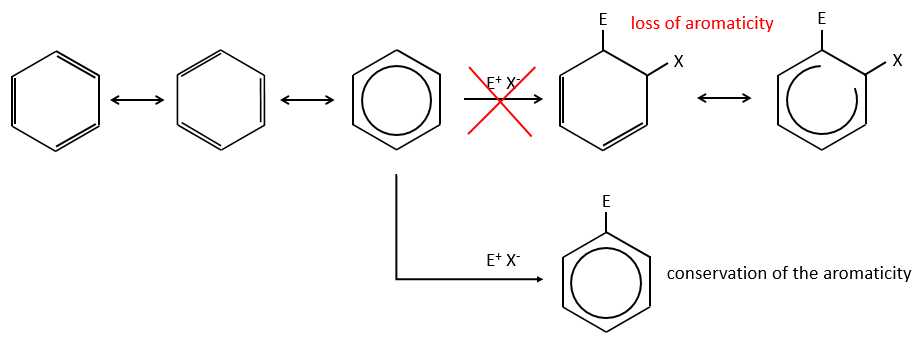
Au lieu de réactions d’addition, on observe des réactions de substitution. Le mécanisme comporte deux étapes.
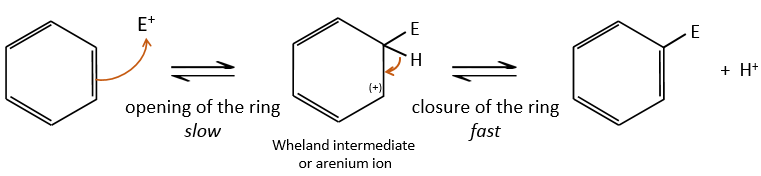
1) la première étape est l’attaque électrophile d’une liaison π sur un électrophile. Comme cette étape implique la perte d’aromaticité du substrat, cette étape est lente et l’étape de détermination de la réaction. Le produit de cette étape est appelé un intermédiaire de Wheland ou un ion arénium.
2) la deuxième étape comprend l’enlèvement d’un proton du cycle pour régénérer l’aromaticité. Cette étape est rapide.
La plupart des électrophiles doivent être activés pour permettre à la première étape de la réaction de se faire. Elle se fait par un acide de Lewis
Halogénation :
FeBr3 et AlCl3 peuvent être utilisés comme acides de Lewis pour lier un atome d’halogène (Br et Cl respectivement) sur un noyau benzénique.
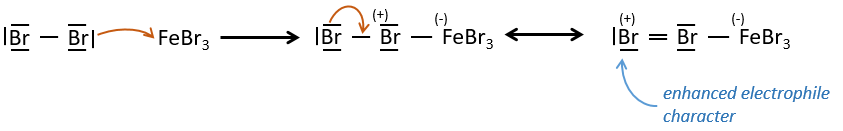
Ils améliorent le caractère électrophile de l’halogène qui peut désormais être attaqué par le substrat aromatique.
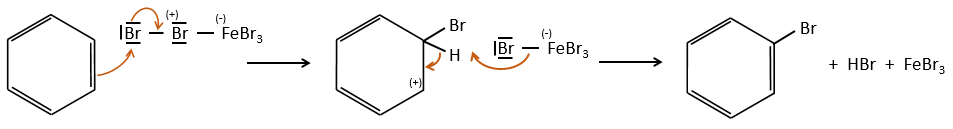
FeBr4– agit comme une base pour prendre un proton et fermer l’anneau.
Dans le tableau suivant nous pouvons trouver des énergies de liaison impliquées dans le processus pour les halogènes. Sur la gauche nous avons les énergies pour les réactifs et dans la colonne du milieu nous avons les énergies pour les produits. L’enthalpie de réaction est représentée sur la colonne de droite.
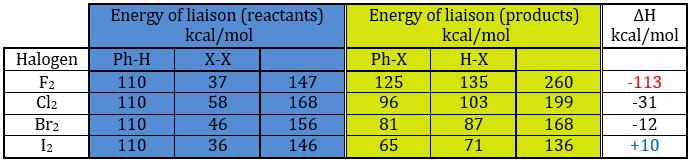
Si nous jetons un coup d’oeil à la variation de l’énergie impliquée par une halogénation, nous voyons que l’enthalpie de réaction est positive pour l’iode. La réaction n’est donc pas faite. La réaction est très exothermique pour F2. En fait cette réaction est explosive, Pour Cl et Br, nous avons besoin d’utiliser des catalyseurs (les acides de Lewis).
La sulfonation et la nitration :
SO3 et NO2+ sont suffisamment électrophiles pour se lier à un cycle aromatique.
La nitration :
Le nitrate peut être réduit de façon sélective pour obtenir une amine
Sulfonation :
L’atome de soufre est électrophile en raison du caractère électrocapteur des oxygènes. Cependant la réaction est réversible en présence d’eau pour former de l’acide sulfurique. Ce processus est exothermique et on devrait garder un oeil sur lui.
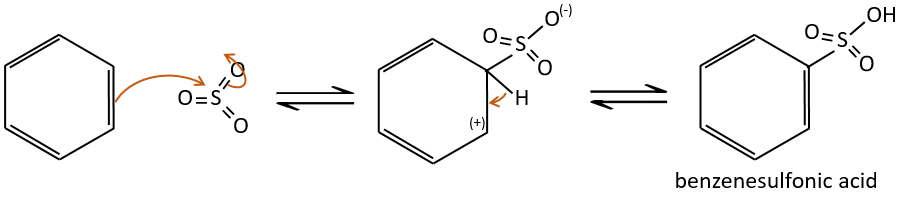
Nous produisons des détergents à partir de l’acide benzènesulfonique.
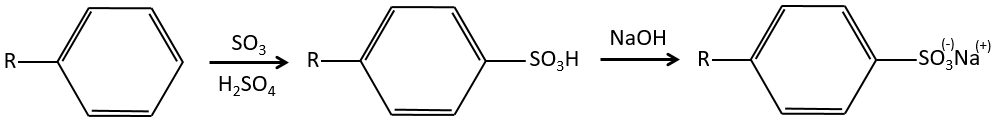
Et à partir de ce point, nous pouvons produire des sulfamides qui sont généralement de bons antibiotiques.
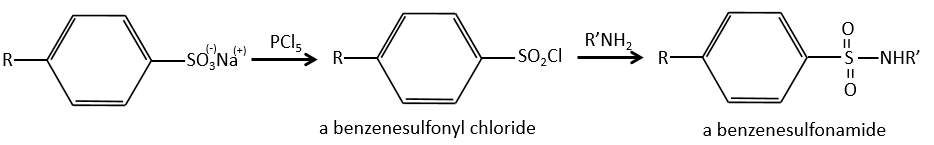
La première de ce genre était le Prontosil (4 – [(2,4-diaminophényl) azo] benzènesulfonamide) en 1932, développé par Domagk chez Bayer, qui a remporté un prix Nobel de médecine pour elle en 1939. Le programme de recherche a été conçu pour rechercher des agents qui pourraient agir comme médicaments antibactériens dans le corps. La découverte et le développement de ce premier sulfamide ont ouvert une nouvelle ère en médecine. Voici d’autres exemples d’agents antibactériens :
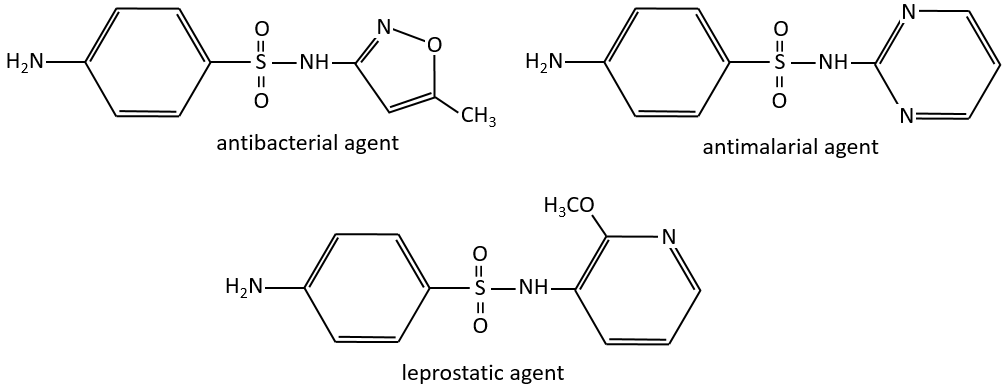
Alkylation de Friedel-Crafts :
Cette réaction, comme son nom l’indique, permet de lier une chaîne sur un cycle aromatique. La chaîne qui doit être ajoutée doit avoir un carbone électrophile. Encore une fois nous avons besoin d’un acide de Lewis pour faire cette réaction. La première étape est l’activation de l’halogénoalcane par l’acide de Lewis.
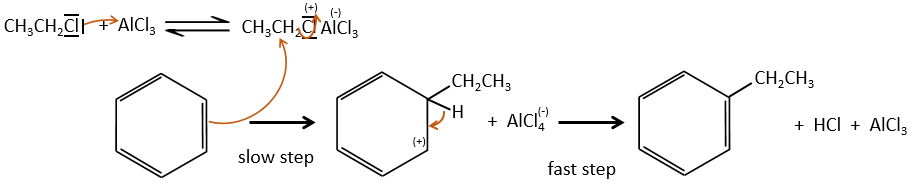
Ensuite le cycle attaque l’alcane, éjectant l’acide de Lewis halogène. Le produit de cette étape est appelé un intermédiaire de Wheland ou un ion arénium. Cette étape est l’étape de détermination de la réaction impliquant l’ouverture de la bague en raison de l’attaque électrophile. Un proton est prise par l’acide de Lewis halogéné pour donner l’aromaticité avant avec la récupération de l’acide de Lewis et la libération d’un acide. Cette étape est rapide par rapport à l’étape précédente. Comme résultat global nous avons ajouté une nouvelle chaîne de carbone sur le noyau phényle.
L’halogène sur la chaîne ajoutée n’est pas obligatoire. Un alcool ou d’autres précurseurs de carbocations peuvent aussi faire le travail.
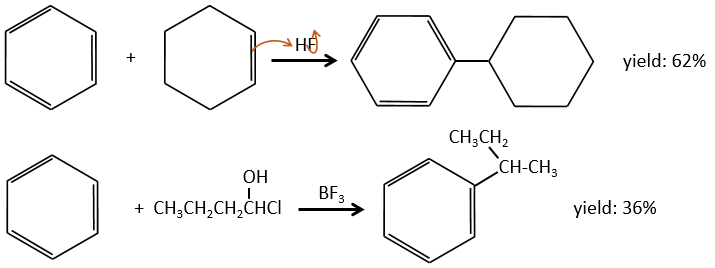
L’alkylation peut être intramoléculaire si elle aboutit à un nouveau cycle.
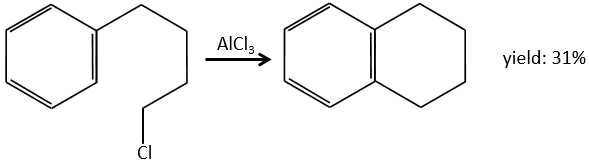
Les limitations de la méthode :
Le groupe alkyle qui a été ajouté au groupe phényle est un groupe donneur inductif. Cela signifie qu’il donne une partie de sa charge au phényle en augmentant sa capacité à attaquer les carbones électrophiles. En conséquence le processus d’alkylation ne cesse pas après l’ajout d’une chaîne si il y a encore de la place sur le phényle pour accueillir de nouvelles chaînes (nous verrons un peu plus tard sur quelle position, quel groupe peut être ajouté).
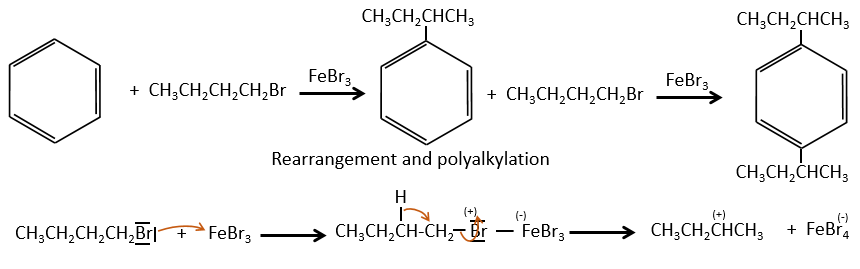
Une seconde limitation est le réarrangement du carbocation (comme d’habitude).
Acylation de Friedel-Crafts
La différence entre l’alkylation et l’acylation est que, pour ce dernier l’électrophile est un cation d’ acylium, conduisant à l’addition de -C = O-R sur le noyau phényle.
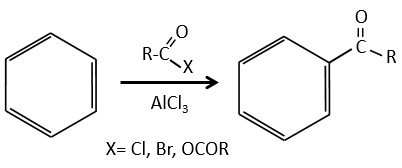
L’ion acylium est obtenu à l’aide d’un acide de Lewis
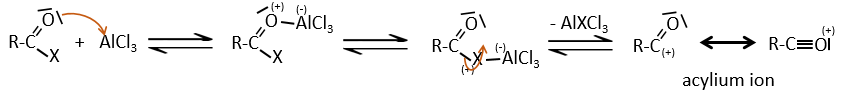
Le reste du processus est identique.
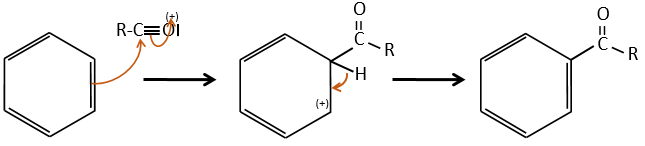
Cependant, il n’y a pas de polyacylation parce que le groupe carbonyle est un capteur inductif et mésomère en prenant des électrons du cycle. Il est possible de réduire la cétone avec la réaction de Clemmensen ou de Wolff-Kishner.
La régiosélectivité :
Un benzène qui porte déjà un groupe peut être attaqué sur 3 positions non équivalentes : ortho, méta et para. Certains groupes peuvent orienter la réaction vers les positions ortho ou para et d’autres groupes vers la position meta.
Effet mésomère :
L’effet mésomère est la possibilité pour un hétéroatome de partager une paire solitaire avec des atomes voisins. Par exemple O et N sont des donateurs mésomères. Ils peuvent stabiliser les carbocations par le partage d’un de leurs paires seules qui contrebalancent la charge positive. Ils améliorent également le caractère nucléophile des liaisons doubles. Nous notons les donateurs mésomères par +M.
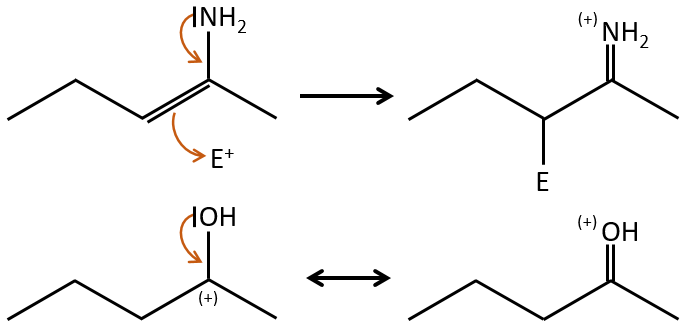
Les groupes peuvent également être accepteurs mésomères et sont notés -M. Par exemple, le groupe NO2 est un accepteur mésomère parce qu’il a une forme de résonance qui peut garder la charge supplémentaire.
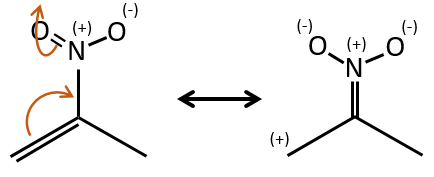
Notez que l’oxygène peut être mésomère donneur ou accepteur en fonction du groupe dans lequel il est et la structure de la molécule.
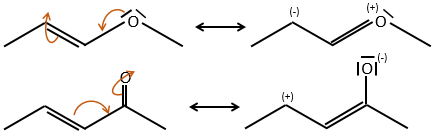
Effet inductif :
Lorsque deux atomes ont une électronégativité différente, il y a un déplacement d’électrons de l’atome moins électronégatif vers l’atome plus électronégatif. Les groupes ou atomes peuvent donc prendre ou donner des électrons depuis ou vers les atomes voisins. Les atomes ou groupes qui prennent les électrons du reste de la molécule sont des accepteurs inductifs et sont notées -I. Ils déstabilisent les carbocations et les charges positives et peuvent augmenter le caractère électrophile de l’atome avec lequel ils sont liés. Par exemple, le carbone d’un carbonyle donne une partie de sa charge à l’oxygène et porte une accusation partielle positif δ+. Un nucléophile est donc enclin à attaquer ce carbone.
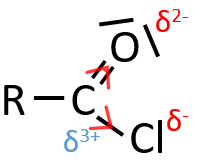
Les atomes qui sont moins électronégatifs que le carbone vont donner une partie de leur charge à la chaîne. Ceux-ci sont appelés les donneurs inductifs et sont notés +I. Le plus simple donneur inductif est H qui est moins électronégatif que le carbone. Cependant H est pris par convention comme l’intensité neutre de l’effet inductif. -CH3 est un donneur inductif parce que l’effet inductif de 3 hydrogènes est transmis à travers la chaîne carbonique (plus de 2-3 carbones). En conséquence les groupes alkyle sont des donneurs inductifs. L’effet est plus fort pour un groupe -C (CH3) 3 que pour -CH3 car le nombre d’atomes d’hydrogène qui peuvent partager leurs électrons est plus élevé.
L’hybridation du carbone est une chose à prendre en compte pour déterminer l’effet inductif. Les électrons dans les orbitales « s » sont plus liés à l’atome que celles des orbitales p. En conséquence l’électronégativité des atomes de carbone sp2 est légèrement supérieure à l’électronégativité des atomes de carbone sp3.
Régiosélectivité en fonction des substituants des cycles aromatiques
Les effets mésomères et inductifs sont cumulatifs et un atome d’azote dans un groupe NH2 est simultanément un donneur mésomère et l’accepteur inductif (+IM). L’effet mésomère prévaut toujours en intensité.
Les donneurs inductifs :
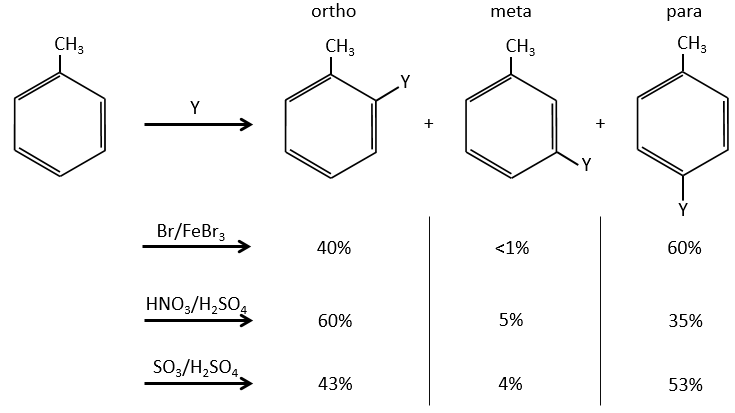
Les groupes qui sont des donneurs d’électrons par hyperconjugaison (ou les donneurs inductifs) activent ou favorisent l’ajout de nouveaux groupes sur le carbone. La substitution est orientée vers les positions ortho et para parce que le carbocation qui en a été fait au cours de la première étape de la substitution peut être stabilisée par le donneur inductif.
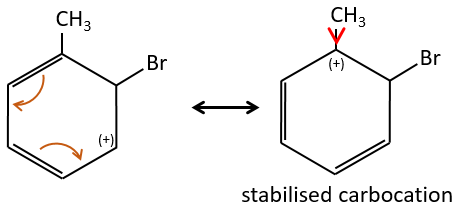
Il est impossible de placer la charge positive à l’endroit du groupe si la substitution devait être faite sur la position méta. Habituellement la position para est favorisée par rapport à la position ortho en raison de l’encombrement stérique. Cependant certains groupes, tels que NO2, semblent montrer plus d’intérêt à la position ortho.
Les capteurs inductifs :
les électrocapteurs inductifs orientent la réaction dans la position méta. La raison est que dans les deux autres positions le carbocation peut être aux pieds du groupe CF3 qui veut prendre plus d’électrons, déstabilisant encore plus le carbocation.
Les donneurs mésomères :
les électrodoneurs mésomères activent et orientent la réaction en ortho/para parce qu’il y a une forme de résonance supplémentaire.
Notez que NH2, ainsi que l’oxygène de l’éther, est un donneur mésomère et un capteur inductif. L’effet mésomère est toujours plus important que l’effet inducteur.
Les capteurs mésomères :
Les groupes qui sont électrocapteurs par résonance orientent la réaction dans la position méta.
Les halogènes :
les halogènes sont désactivants mais orientent la réaction en ortho et para.
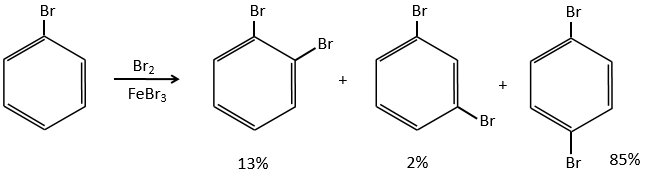
L’encombrement stérique ici est très important et explique la différence de population entre les réactions ortho et para.
S’il y avait deux groupes sur le cycle phényle les effets sont additifs et la substitution est faite sur les positions les plus activées/moins désactivées.
Les deux groupes méthyle du xylène (diméthylbenzène) indiqués ci-dessous sont activants et orientent dans ortho/para. Comme les deux sont des groupes d’activation ce réactif est plus réactif que le toluène. Les positions 2, 4 et 6 sont ainsi favorisées. Les positions 4 et 6 sont légèrement plus probables car il y a moins d’encombrement stérique. Dans le cas du xylène les fentes sont à peu près équivalentes.
Dans le cas des groupes de désactivation la même réflexion est faite. Le COOH oriente en position méta et la position 5 est la position la moins désactivée.
La substitution nucléophile :
La substitution nucléophile sur des noyaux aromatiques est plus lente que la substitution électrophile et les substitutions sur des carbones sp3.
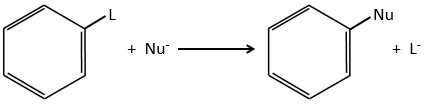
La raison en est que le cycle est déjà plein d’électrons. En outre un carbone sp2 est plus électronégatif que le carbone sp3. Il est donc difficile d’ajouter un nucléophile (qui aime les charges positives) sur un groupe phényle et la présence d’un groupe de capteur sur le phényle est nécessaire pour stabiliser le carbanion. Le groupe partant doit être un bon.
Plusieurs mécanismes sont possibles pour ajouter un nucléophile sur un substrat aromatique. L’un d’eux est l’addition-élimination.
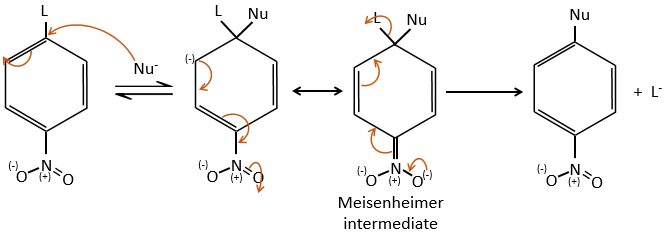
Le groupe d’activation (ici NO2) est nécessaire pour stabiliser la charge négative du carbanion formé lors de la première étape de la réaction qui est la plus lente. Le complexe intermédiaire est appelé complexe intermédiaire de Meisenheimer.
La séquence de réactivité halogène (comme groupe partant) est opposée à celle de SN2. Sur une chaîne aliphatique le clivage de la liaison C-X est faite au cours de l’étape déterminant de la SN2. Sur un cycle aromatique le clivage ne se produit pas au cours de l’étape déterminante. En outre les halogènes prennent des électrons du cycle et les petits halogènes sont donc plus réactifs que les grands. En conséquence nous avons les séquences de réactivité suivantes :
Aliphatique : F<<Cl<Br<I
Aromatique : F>>Cl>Br>I
Un atome hétérogène dans le cycle (O ou N) peut jouer le rôle du groupe de capteur.
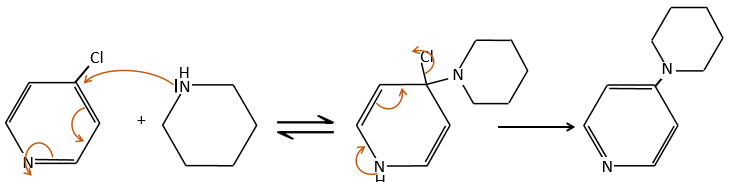
Une application de ce mécanisme est la détermination de l’acide aminé terminal des peptides. Le peptide réagit avec un composé aromatique par l’intermédiaire de son groupe amine terminal.
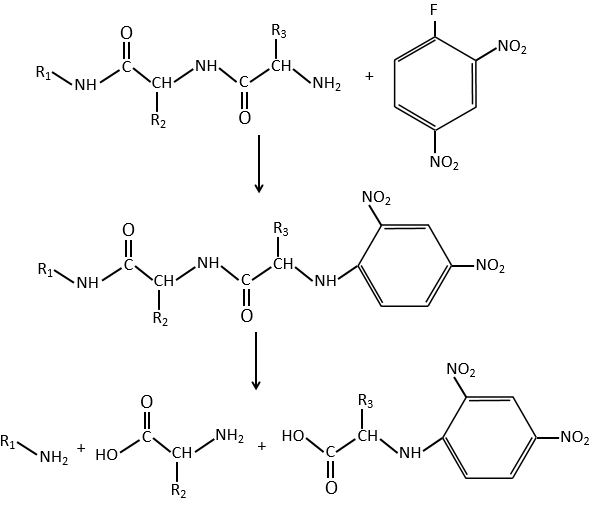
Une fois que les deux substrats sont liés ensemble nous hydrolysons les liaisons peptidiques (cf les amides). Tous les acides aminés sont maintenant séparés mais seulement l’un d’eux est lié au groupe aromatique.
Le mécanisme SN1
Ce mécanisme est surtout utilisé pour produire des sels d’arènediazonium. Cette espèce est obtenue à partir de l’aniline C6H7N avec NaNO2 dans un environnement acide.
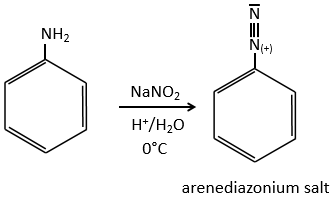
Le mécanisme est le suivant :
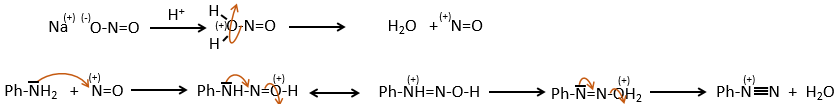
Les sels d’arènediazonium sont stables à basse température et perdent N2 à des températures plus élevées, ce qui libère un cation phényle qui peut réagir avec un nucléophile.
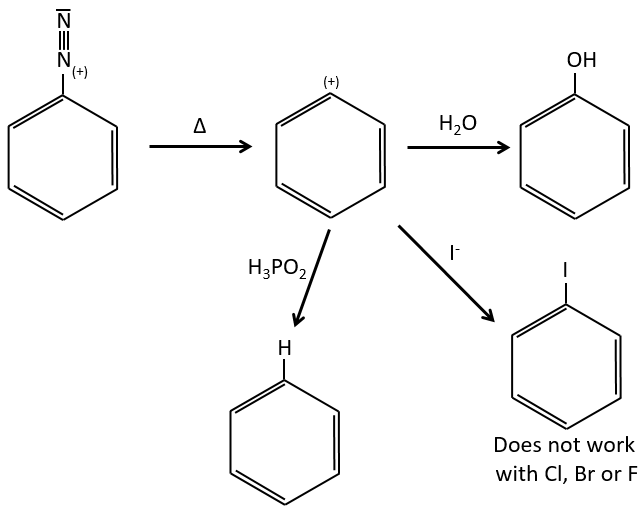
Les halogènes autres que l’iode ne donnent pas de bons résultats en raison de réactions secondaires. Pour ajouter Cl, Br ou F sur un cycle aromatique, on utilise la réaction de Sandmeyer en utilisant des sels de cuivre tels que CuCl, CuBr ou CuCN. Le mécanisme est un peu plus complexe et fait intervenir des radicaux.
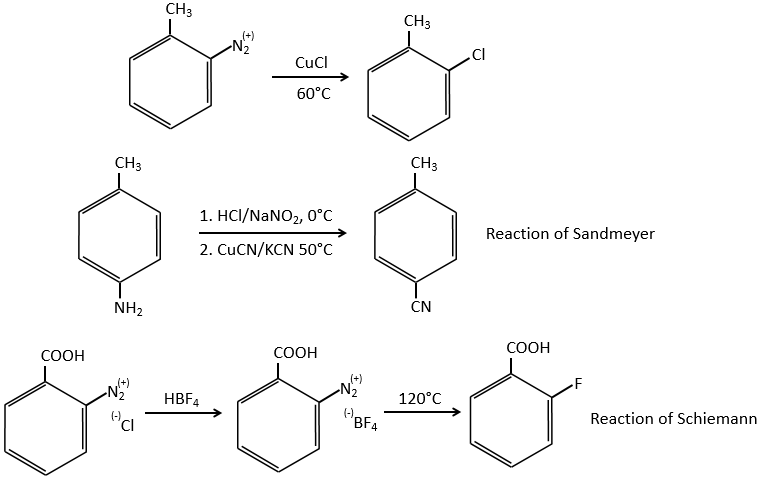
Le mécanisme comportant un benzyne :
Normalement les halogenoarenes ne peuvent pas faire des réactions SN2 ou SN1. Toutefois, dans des conditions très dures de température et de pH, il est possible de forcer pour faire de telles réactions.
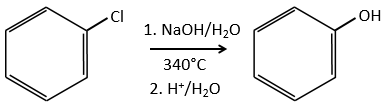
Le mécanisme implique une triple liaison dans le cycle, les espèces étant appelée benzyne, l’existence de cet intermédiaire a été montrée par un marquage isotopique. Le C lié à l’halogène est un isotope et nous observons un mélange racémique en tant que produit: