We will now discuss the types of reactions that can be performed on organic compounds. In this first year lessons, we will only focus on one of the most used reactions: the nucleophilic substitution (SN).
A substitution reaction is indeed the base of the organic chemistry: its goal is to substitute one group of a given molecule by a desired other one. Other important types of reactions are the addition and the elimination (they will be seen in the second year courses most probably).

At the end, we can, in theory, do whatever we want to organic compounds but it is obviously not as easy as said. To this point, you only had the explanation of half of the name of the nucleophilic substitution.
The nucleophile notion is also a very important in organic chemistry. A nucleophile is a compound, rich in electrons, that is attracted by nucleus (poor in electrons) and can share a pair of its electrons to form a liaison. By opposition, an electrophile is a compound, poor in electrons, that is attracted by electrons. On the other hand, acidity as we have seen it previously is not that important. Acidity is important in aqueous solutions and organic compounds do not dissolve well in aqueous solution but are in two separated phases. In aqueous solutions, species are dissociated and in organic solutions, they are not. The separation method takes advantage of that to purify or separate compounds.
During a nucleophilic substitution, a nucleophile attacks a target molecule to substitute a group, called the leaving group (LG). The target of the nucleophile is a carbon atom poor in electrons because it wears a group of an atom which is more electronegative. It limits the possibilities of targets to halogens and groups with O or N (not nitro groups because the azote has a positive charge there). In the case of SN, the leaving group takes a pair of electrons with itself.
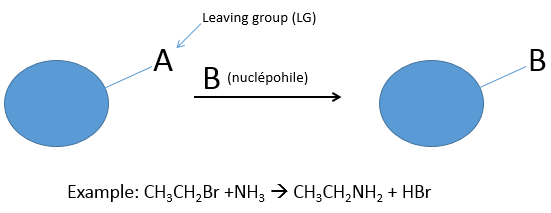
The leaving group is a nucleophile itself and for the reaction to be made, the attacking nucleophile has to be a stronger nucleophile than the leaving group. Several parameters are determining the nucleophile strength (or nucleophilicity).
Charge of the nucleophile
First of all, a nucleophile has not to be charged. Possessing a lone pair of electrons is enough. H2O, NH3, CH3OH are all nucleophiles thanks to their free pairs of electrons (on the O and on the N). However, their negatively charged equivalent is a stronger nucleophile.
![]()
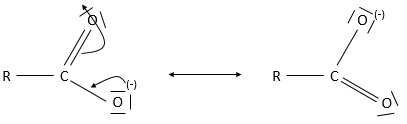
Either the charge or the free pair of electrons have to be available for the reaction. If the charge (or the lone pair) is delocalized, the nucleophilicity decreases significantly. Despite all their lone pairs and their charge, carboxylates are not good nucleophiles because the charge can be displaced. We say that a carboxylate is stabilized by resonance because this compound has a smaller tendency to react with other compounds because of its two resonance forms.
Basicity
Even if the acidity is not so relevant in organic solutions, there is a good relation between the basicity of a compound and its nucleophile strength.
Electronegativity
The nucleophilicity decreases with the electronegativity of the atom wearing the charge/lone pair. Remember that electronegativity is the ability of the atom to keep its electrons. As a result, the electronegative atom won’t share its electrons to form a liaison.
Polarisability
Nucleophilicity decreases with the polarisability of the atom. While it is counterintuitive with regards to the reaction, this relation is due to the solvation of the nucleophile. In protic solvents, the nucleophile species are surrounded by solvent molecule, forming hydrogen liaisons with the lone pairs of the nucleophile. Interestingly, the solvation is stronger for small molecules than for larger ones because the charge density is larger for smaller molecules.
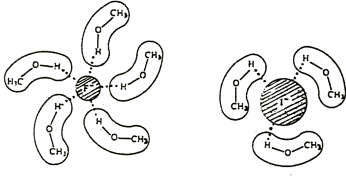
In aprotic solvents, this effect does not exist and the polarisability favours nucleophilicity.
Mechanisms for nucleophilic substitutions
There are 3 possible mechanisms for a substitution:
- First the attack of the substrate by the nucleophile, then the ejection of the leaving group
- Simultaneous attack of the nucleophile and departure of the leaving group
- First the departure of the leaving group and then the attack by the nucleophile.
The first mechanism is quickly ignored because the first step leads to a pentavalent carbon atom. The two other mechanisms exists and are in concurrence. In some conditions, both can occurs in the same time. We will now see the mechanisms in detail and see the differences between them.
SN2: Bimolecular Nucleophilic Substitution
SN2 is the second mechanism of the list: simultaneous attack of the nucleophile and departure of the leaving group. The whole process is done in one step and the speed of the reaction depends on the concentrations of the two reactants (the substrate and the nucleophile). It is the reason why this mechanism is called bimolecular.
Our first step is to determine where the nucleophile attacks on the substrate. Considering the substitution of a chlorine by an alcohol on a given molecule (X, Y and Z being the rest of the molecule), the nucleophile (OH–) has two options:
- Attack by the side of the chlorine
- or at by the other side
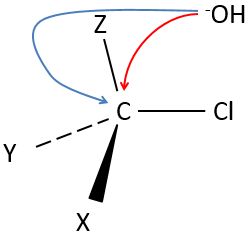
Both sides give the same result in its composition, but the connectivity differs.
Attack by the side of the leaving group (called frontal attack)
To be simultaneous, the liaison between the substrate and the leaving group loosens while the liaison between the substrate and the nucleophile forms itself. It gives place to a transitory complex to obtain next the substituted alcohol.

The connectivity of the formed molecule is identical as the initial one: X, Y and Z do not change of position during the process.
Attack by the other side (called dorsal attack)
Again, a transitory complex is formed. This complex shows differences with regards to the transitory complex observed for a frontal attack: the conformation of the molecule changes during the process. The liaisons between X, Y and Z with C are in a single plane, perpendicular to the “liaisons” with OH and Cl.
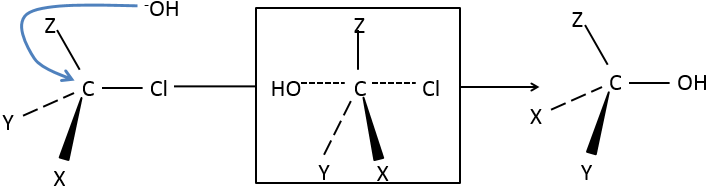
As a result, the position of X and Y are inverted. The products of the frontal and dorsal substitutions are chiral products (mirror images from one each other).
We can thus easily determine which results are obtained using the optical activity of each enantiomer. It showed that only the dorsal substitution is done. The frontal attack does not occur.
There are two consequences:
- there is an inversion of configuration after a SN2
- steric hindrance slows or blocks the reaction. The size of X, Y and Z plays thus an important role in the speed of the reaction. For example, the SN2 goes 145x faster for CH3Br than for CH3CH2Br, which goes itself 128x faster than (CH3)2
SN1: Monomolecular nucleophilic substitution
In this mechanism, the departure of the leaving group acts first and then the nucleophile attacks the substrate.
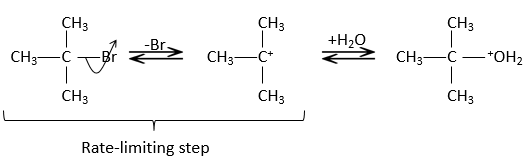
There are thus two steps in this mechanism. The global speed of the reaction is given by the slowest step ‘called the rate-limiting or rate-determining step), which is the departure of the leaving group. Indeed, it implies the breaking of a liaison and a separation of charges. A protic solvent helps to separate the carbocation and the leaving group, through solvation of the charged species. The second step is much faster: the nucleophile is attracted by this carbon wearing the positive charge.
The limiting step is thus the first step of the substitution, where the leaving group is dissociated of the substrate. This reaction implies only one reactant, the nucleophile acting only at the second step of the substitution. The name of SN1, monomolecular nucleophilic substitution comes from there. The kinetics depend then only on the concentration of the substrate, contrarily to the SN2 where the concentration of the nucleophile has an influence on the speed of reaction. A way to determine if a substitution reaction is SN1 or SN2 is to perform a kinetics analysis, determining if the concentration of the nucleophile has or has no influence on the speed of reaction.
To have a SN1, the positively charged carbon, called carbocation, has to be stabilized.
Carbocation
A carbocation is a carbon wearing a positive charge. It has three liaisons in a single plane and an empty orbital perpendicular to this plane.
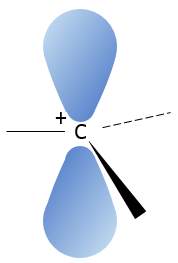
That geometric configuration stabilizes the carbocation with optimized angles between liaisons (nb: plane or quasi plane structure has to be obtained) but also by hyperconjugation. Hyperconjugation is the interaction of the electrons in a sigma bond (usually C–H or C–C) with an adjacent empty (or partially filled) non-bonding p-orbital (the case of carbocations), antibonding σ or π orbital, or filled π orbital, to give an extended molecular orbital that increases the stability of the system. In other words, the liaisons of adjacent carbons of the carbocations stabilize the carbocations when correctly oriented.
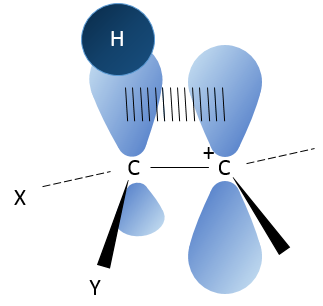
In the picture above, one can see the hyperconjugation of a sp liaison between C-H with the empty p orbital of the carbocation.
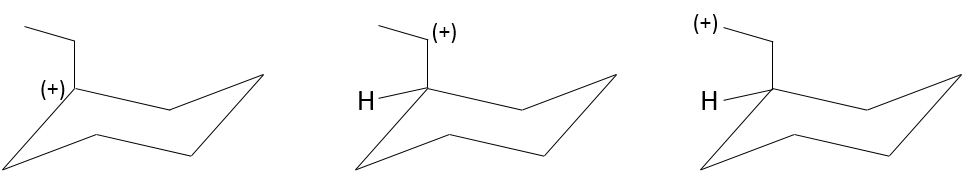
As a consequence, substituted carbocations are stabilized through hyperconjugation. The sequence of stability for carbocations is thus:
![]()
As you have maybe already understand, one parameter to determine if the mechanism of substitution is a SN1 or a SN2 is the substitution of the carbon wearing the leaving group. Trisubstituted carbons lead to SN1 and monosubsituted or unsubstituted carbons lead to SN2. With bisubstituted carbons, both mechanisms can take place, and are pretty slow.
Adjacent π-liaisons also stabilize carbocations through (hyper)conjugation of orbitals and through resonance.
![]()
Let’s come back to the mechanism of SN1. Carbocations are plane and can be attacked by both sides of the plane. The proportions of attack on each side are equal except if there is a difference of steric hindrance for one side.
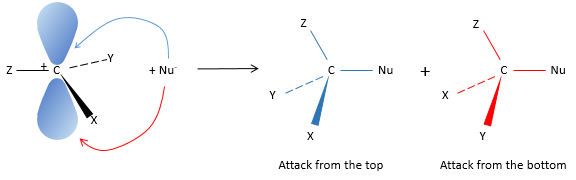
As a result of the SN1, we obtain a racemic melange of two enantiomers.
However, we can sometimes obtain very different and unexpected products from a SN1. For example,
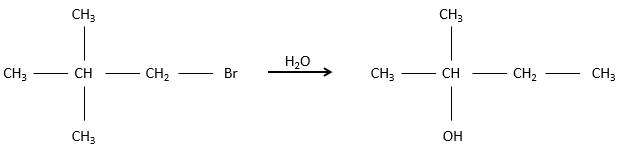
This product is quite unexpected: the leaving group was not substituted by the nucleophile but by a methyl while the nucleophile took the place of a methyl not even on the same carbon than the leaving group. The reason why such a product is obtained is that the carbocation produced during the SN1 was not the most stable carbocation that the molecule could have. Through a not totally understood mechanism (called Wagner-Meerwein mechanism), a migrant group (a methyl or a Hydrogen) is displaced to obtain a better carbocation:
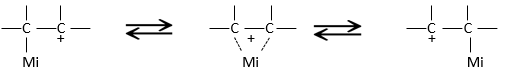
If a methyl and a Hydrogen were on the adjacent carbon, the hydrogen is displaced because it leads to a better carbocation

Only then the nucleophile attacks the carbocation.
SN1 vs SN2
The mechanisms themselves are not complicated but it may be hard to determine which mechanism takes place. We can resume the parameters influencing the substitution mechanism as follow:
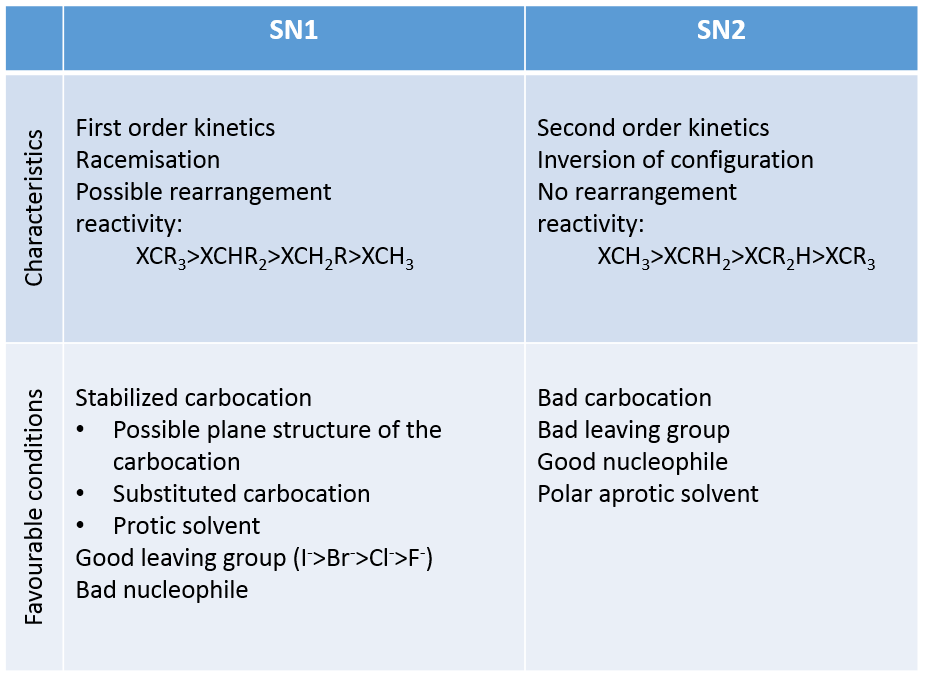
As it was said at the beginning of this chapter, nucleophilic substitutions is one of the most important reaction in organic chemistry. Basically, you will want to substitute a group to extend a chain, to place a desired group on the substrate and to protect a group.
Other substitutions than SN1 and SN2 exist but we won’t talk about them now. They are much less common.
Exercises
1. Sort those compounds from the best to the worst nucleophile:
NaOH CH3COOH N+(CH3)4Cl– SH2 NaI
C4H5N N(CH3)3 CH3OH H2O NH3
2. Which product and mechanism is used for the following reactions:
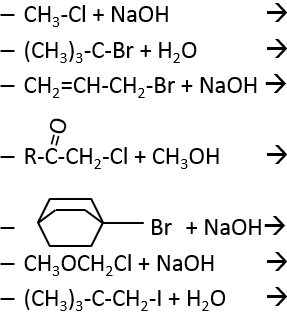
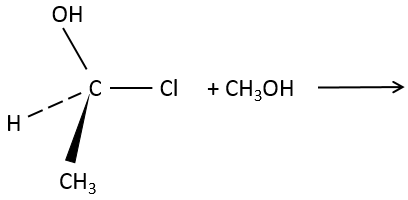
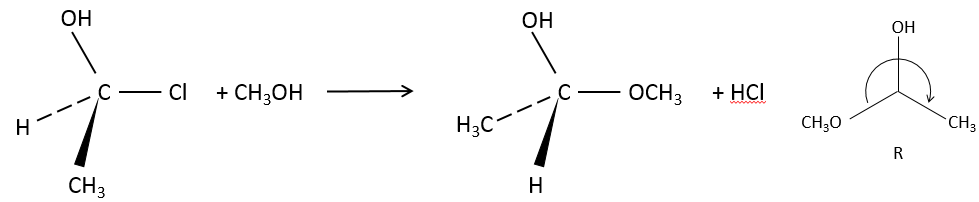
3. What is the product and its configuration for the following reaction:
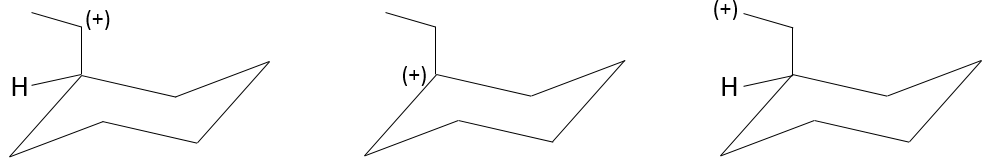
4. Sort those carbocations by stability
Answers
- NaI>NaOH>SH2>NH3>H2O>CH3OH>CH3COOH>N(CH3)3>C4H5N>N+(CH3)4Cl–.
The anions are the best nucleophiles. I- is better than OH- because Iodine has a very high polarisability. SH2 and NH3 can both be compared to H2O. NH3 is a better nucleophile than H2O because N is on the left of O on the Mendeleev table (i.e. more basic). SH2 is a better nucleophile than H2O because S is under O on the Mendeleev table (i.e. more polarisable). Between SH2 and NH3, SH2 is the best. Next come H2O and CH3OH which are weak nucleophiles. Here the size matters. The carboxylic acid is very weak because of its resonance forms. The tertiary amine is very weak as well because the lone pair is sterically hindered by the methyl groups. In the next group, the lone pair of the azote takes part in the aromaticity of the cycle. The last group is not at all nucleophilic: positively charged, hindered and no lone pair of electrons.
2. The reactions are the following:
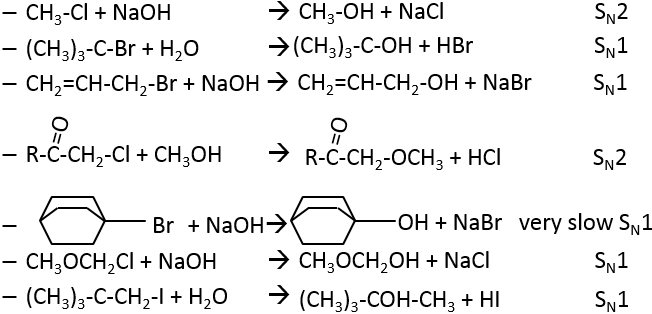
3. The product is the R enantiomer:
4.From the most stable to the less stable: