Chapter 5 : asymmetric synthesis – formation of optically-pure compounds
There are two ways to obtain such compounds. The first method is called the resolution in which the reactants we are starting with are either racemic or achiral. We separate the enantiomers after the production of both enantiomers. This method is vastly used in industries even if it doubles the volumes. They try to use the inactive enantiomer in another process. During a diastereoisomeric resolution, the separation of the enantiomers is made by the addition of an optically pure enantiomer called the resolution agent.
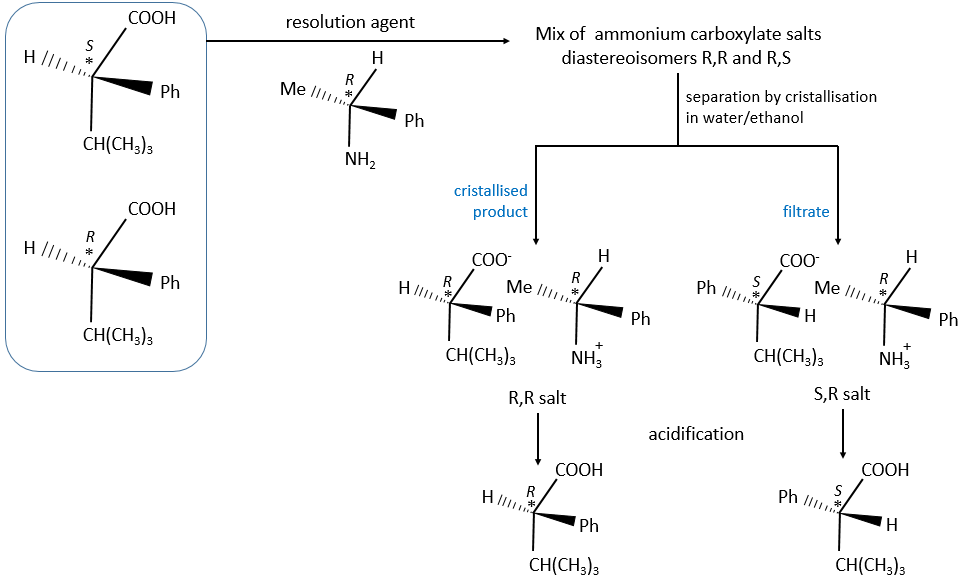
The formed salt is a racemic melange of diaseteoisomers that can now be separated.
The separation of the enantiomers can also be done via the kinetic splitting that is based on the difference of reaction speed of the enantiomer with a given chiral reactant.
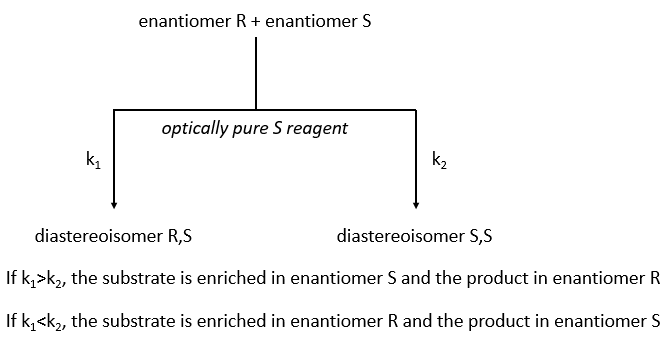
The speed of reaction has to be very different for the two enantiomers. If we put 0.5 equivalent of the chiral reactant, the enantiomer that reacts faster will from the diastereoisomer and the other enantiomer remains intact (ideal case). The conversion ratio is the proportion of the intact enantiomer over the proportion of the formerd diastereoisomer.
The second way is to directly produce the chiral species. This technique evolved over the years and began with the use of a chiral substrate. Around 1980 a chiral auxiliary was used. In this decade, they also used chiral reactants and after 1990 we started to use chiral catalysts. We will describe those techniques soon. First we will talk about the chiral pool.
The chiral pool
The chiral pool refers to natural compound that are optically active and can be produced at large scale (and thus are cheap). It can be aminoacids, peptides, metabolites, etc. We can use those compounds as starting reactants to obtain a chiral final product. The stereocentres already present on the natural compound will control the stereoselectivity of the reactions and lead to one single enantiomer. We talk about a hemisynthesis (because the starting reactant has not be chemically produced).
Amino-acids
At the exception of the glycine, the 20 amino-acids are chiral and available with a good ee (enantiomeric excess). They are bifunctional and levogyre but an enzymatic method can reverse their configuration.
Hydroxy-acids
They wear one or more OH and COOH and can have several stereocentres.
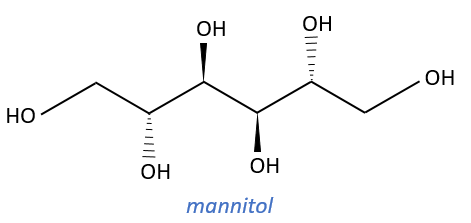
Carbohydrates
They are very cheap but we have to modify them and to protect some groups to use them correctly.
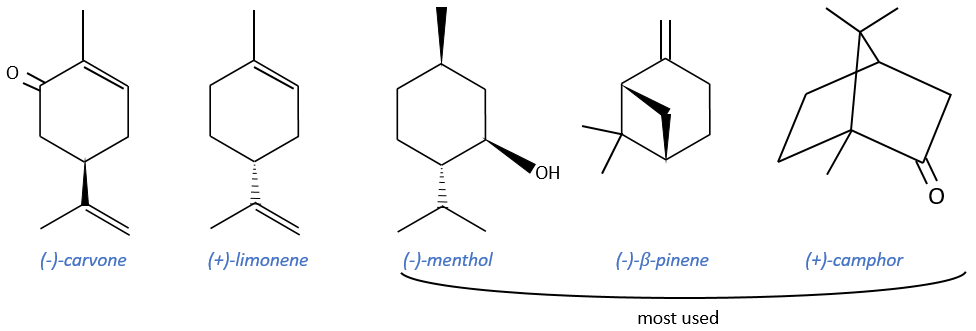
Terpenes
Most of them are used as precursors of agent of resolution or as ligands during asymmetric catalysis but they can also be used as starting reactant of other terpenes. They are cheap and available in large quantities. Their cyclic structure allows a better control of the strereoselectivity. However, they don’t have a lot of functional groups and their optic purity is not always very good.
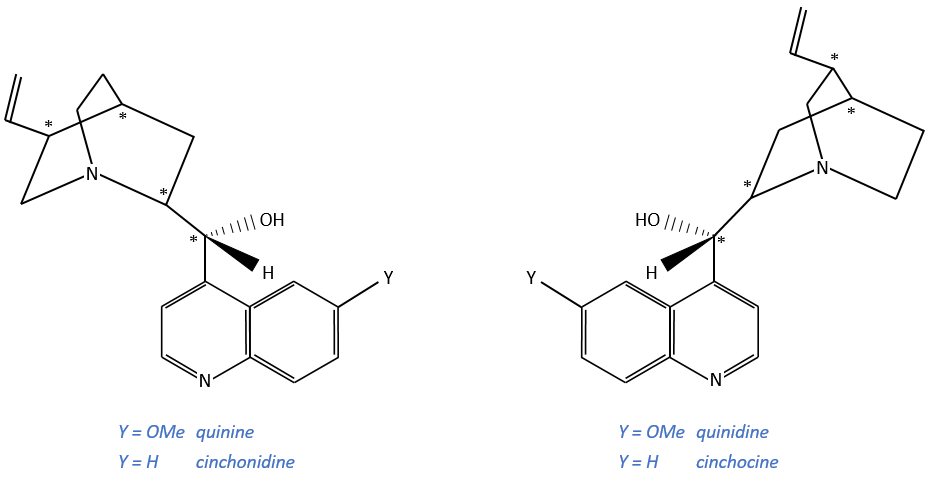
Alcaloïdes
These secondary metabolites possess a nitrogen aromatic and are used as chiral catalysts or agents of resolution.
Chapter 4 : asymmetric synthesis – analytical methods of determination of the enantiomeric excess
A chiral agent is always necessary to differentiate the enantiomers. The most used methods to determine the ee are
- the GC with a chiral stationnary phase,
- HPLC with a chiral stationnary phase,
- NMR with chiral lanthanides.
Gazeous chromatography
The stationnary phase of the column is chiral and only composed of one enantiomer (for instance the R configuration). When a racemic melange passes through the column, the enantiomers R’ and S’ have a different interaction with the chiral stationnary phase with which they form diastereoisomeric complexes.
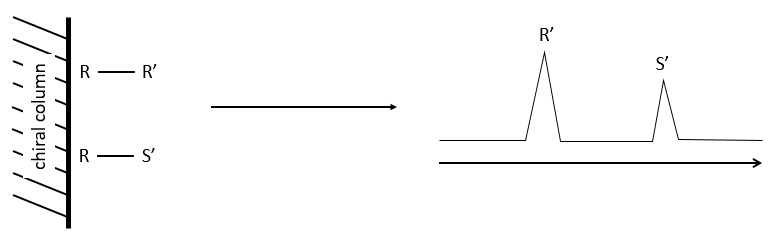
The main interactions can be H bonds, sterical interactions or dipole-dipole interactions. As the interactions have not the same strength, the enantiomers R’ and S’ have a different retention time and exit the column separately, giving two peaks.
The advantages of this method is that one the condition of experimentation are setted up, the analysis is fast and has a high sensibility. It can detect an enantiomer even if its proportion in the solution is about 0.5%. Moreover, the analysis separates the species composing the sample and we can select the portion of the solution that we want to analyse next.
When we use this method, the racemic melange has first to be tested to be sure that the enantiomers can be separated and that the ratio of the areas of the peaks is 50:50.
NMR
This technique is not as sensible as the GC but it is very fast and cheap.
Normally, enantiomers have the same NMR spectrum because the interatomic distances are identical for the two enantiomers. Diastereoisomers have spectra that can be distinguished. To distinguish two enaintiomers we will thus transform them into diastereoisomers.
![]()
To do so, we use two types of chiral reactants.
- chiral agent of derivatisation
- chiral lanthanides
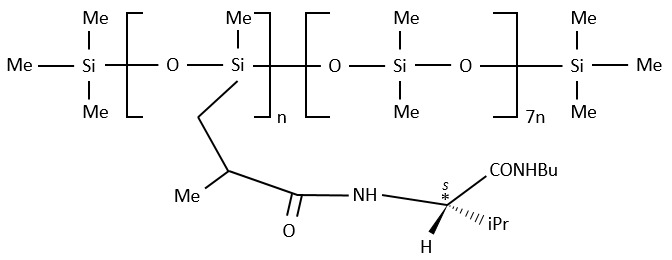
A chiral agent of derivatisation is a chiral compound optically pure that interacts with the enantiomers. The most used one is the Mosher’s acid (α-methoxy-α-trifluoromethylphenylacetic acid (MTPA)).
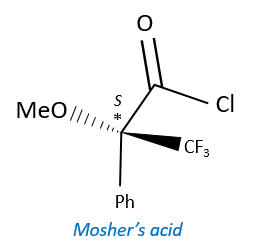
This reactant forms a covalent liaison with the enantiomer to form diastereoisomers with an important difference of chemical displacement. The ratio between the areas gives the enantiomeric excess.
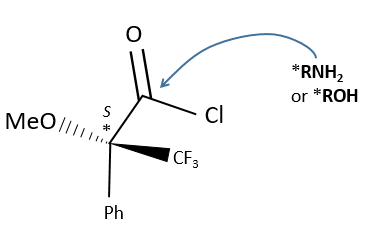
The method with chiral lanthanides is more used than the method with a chiral agent of derivation.
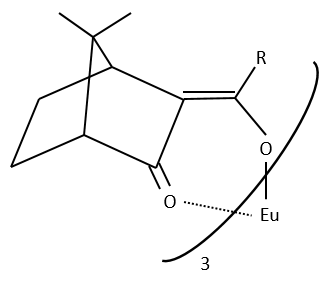
Lanthanides allow a widening of the NMR spectrum by the displacement of the signals towards the low fields. The salt of lanthanide is added bit by bit to the sample to observe the separation of the signals. They form a complex with the enantiomer without covalent liaison but still give an important separation of chemical displacement.
The disadvantages of this method is that it takes more time (the salt is added slowly) and that the salt is expensive.
Chapter 3 : asymmetric synthesis – stereoisomery
Editor’s note: It is a good exercise to determine the configuration (R or S) of each chiral centre presented in this course.
Some molecules have the same composition in terms of atoms but differ by their placement. Those molecules are called conformers (of configuration). Amongst them we distinguish enantiomers and diastereoisomers. The distinction was already explained in a previous section and we will explain it here too. Despite one slight difference of arrangement, the activity of two enantiomers can often differ signifficantly, especially in biology where the damages can be important. Pharmaceutical industries have to separate the enantiomers in the medecines or prove that the other enantiomer is truely inactive. Separation methods exist to isolate one of the enantiomer but it is one more step in the process and there is still 50% of product that is simply lost. For that reason, the field of asymmetric synthesis developped quickly in the current of the 1980’s.
Examples of problems due to the enantiomer
A melange of enantiomers can be less active for antagonism reasons: one enantiomer blocks the activity of the other enantiomer. For instance, the R pheromone of japanese cockroaches becomes totally inactive if the isomer S is present (the notion of R,S, (+) and (-) will be explained soon), even in a proportion of 1%. The S isomer can fix itself on the same receptor than the R isomer but it is inactive. Once it is on the receptor is doesn’t leave, making the R isomer inactive.
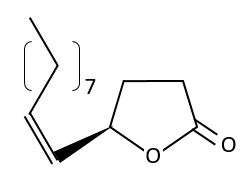
The use of a melange can be dramatic. Thalidomide is a medecine that treat the symptoms of sleeping troubles and of depression. The molecule is active even in presence of its isomer but the isomer causes damages to the phoetus of pregnant women. Disformed babies were born because the enantiomers were not separated at this time.
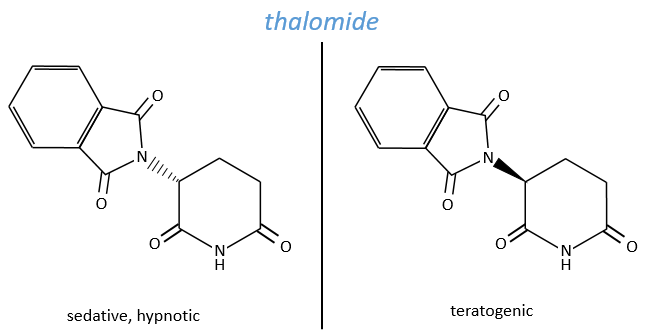
On the figures above, we use three types of lines to represent liaisons:
- a full line indicates either that the liaison is in the plane of the sheet or that its direction outside this plane has no interest,
- black triangles mean that the liaison goes out from the plane of the sheet (point side of the triangle) towards the reader (base of the triangle). It can be seen as a full line we see closer and closer,
- parallels lines in the shape of a triangle is the opposite of one black triangle: the liaison goes from the plane of the sheet (small side) to away from the reader (large side).
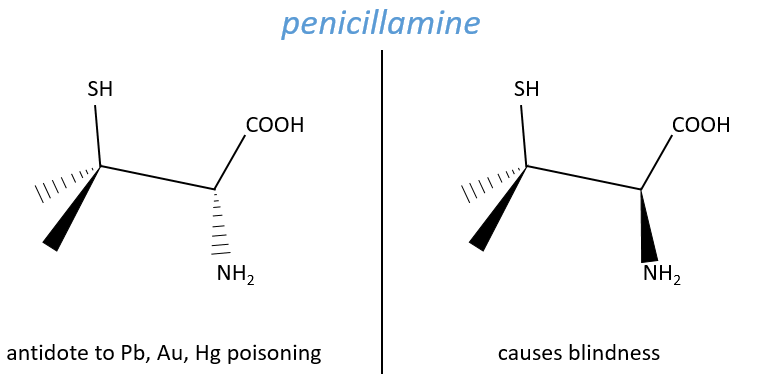
Another example is the penicillamine. One isomer is an antidote to poisoning by Pb, Au or Hg. The enantiomer can make you blind.
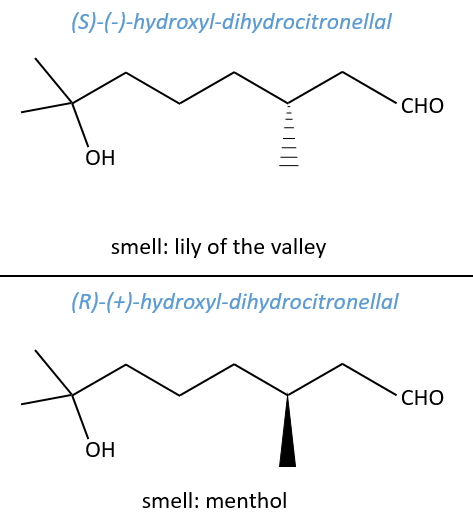
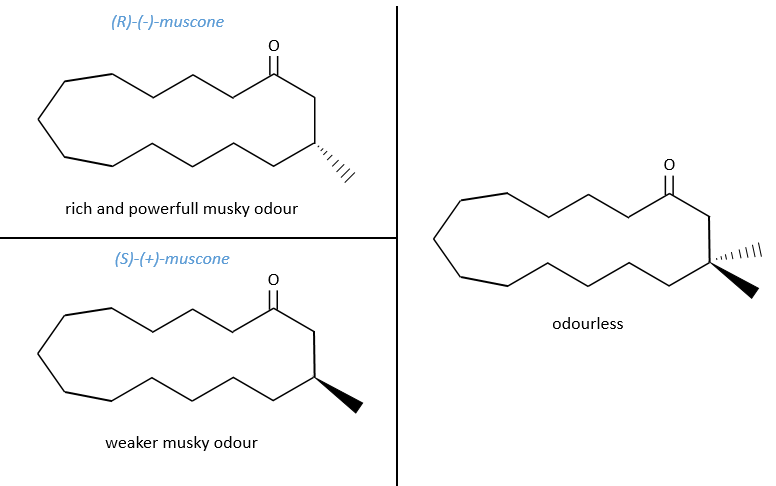
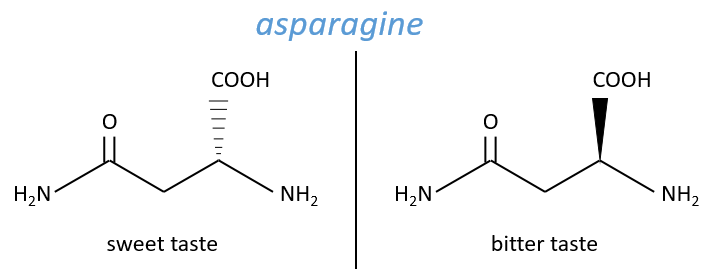
No need to say that it is better to avoid any melange of the two enantiomers. Eniantiomers can also show big difference in taste or smell because they will activate different receptors in our nose/on our tongue.
Reminder on the stereoisomery
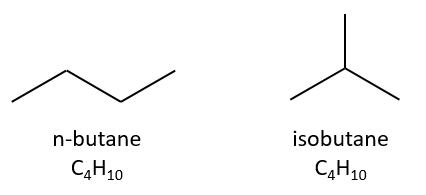
Isomers are compounds of same composition but with a different structure. Isomers of constitution have not the same arrangement of atoms. A simple example is the n-butane and the isobutane.
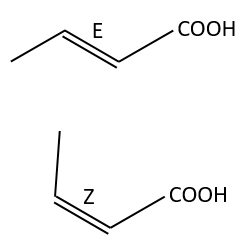
Amongst the isomers, we distinguish stereoisomers. In stereoisomers, the sequence of atoms is identical but the spacial arrangement of the atoms is different. Stereoisomers of configuration can be separated and isolated. To pass from one stereoisomer of configuration to another one, one liaison has to be broken. For instance, the configuration around a C=C liaison can be Z or E, but we have to break the π liaison to go from one to the otherone.
Stereoisomers of conformation, or conformers, are different arrangements of the same molecule obtained by the rotation of one (or more) σ liaison. There is a barrier of energy for this rotation, which can be huge if the rotation is made difficult by the presence of a sterically hindred substituent. Moreover the rotation depends on the temperature. An simple example is the chair-boat conformers of the hexane.
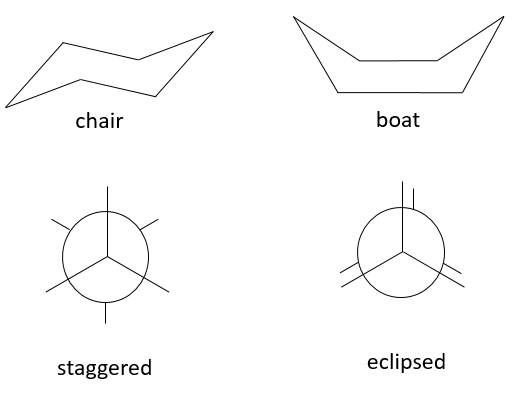
The chair conformation is favoured by 6kCal/mol because all the substituents are in staggered position while they are all in eclipsed position in the boat conformer, leading to a bigger sterical hindrance. There is an equilibrium between the two conformers which is completely displaced towards the chair conformation at ambiant conformation. If we decrease the temperature we can observe both conformers and even separate them. The barrier of energy depends on the substituents of the hexane and the compound is more stable when big substituents are on an equatorial spot because the steric hindrance is smaller there. On the boat conformation, the steric hindrance is always more important because all positions are eclipsed.
Sometimes the rotation of one liaison is so much hindered that the life time of the isomer is long enough to be considered as a stereoisomer instead of as a conformer. As a general rule, we say that we are in presence of conformers if ∆G¹<22.6kCal/mol. If it is larger, then we are in presence of stereoisomers (atropoisomers: denial by steric hindrance). This value corresponds to a life-time of approximatively 1 hour at 25°C.
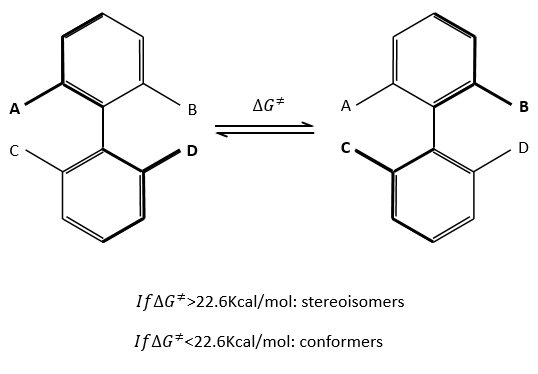
Binaftines are composed of two benzenes bound by one σ liaison. If there is no substituent on the aromatic cycles, the molecule is plane. The liaison between the cycles is not completely one σ liaison as all the π liaisons are conjugated. If there is one or more substituent on A, B, C or D, the steric hindrance doesn’t allow the planarity of the molecule and it becomes very difficult for the σ liaison to make a complete rotation. For instance, the binaftol cannot turn at ambiant temperature.
Chirality
The chirality is the property of an object not to be superimposable to its image in a mirror. The usual example of chiral objects is our hands. The left hand is the mirror image of our right hand but we cannot superimpose them. Indeed, if our palms are in front of us, the fingers are not at the same place. For instance thumbs are not at the same side of the hand. Even if we rotate one hand by 180°, fingers are at the same place but not in the same direction. The word itself, chirality, is derived from the greek word for hand. As a general rule, if a molecule has no plane or centre of symmetry, this molecule is chiral. Yet, there can be an axis of symmetry in achiral molecules.
The stereogenic unity responsible for the chirality of the molecule can be
- an atom,
- an axis
- a plane
- the helicity of the molecule
There are thus 4 main types of chiral molecules.
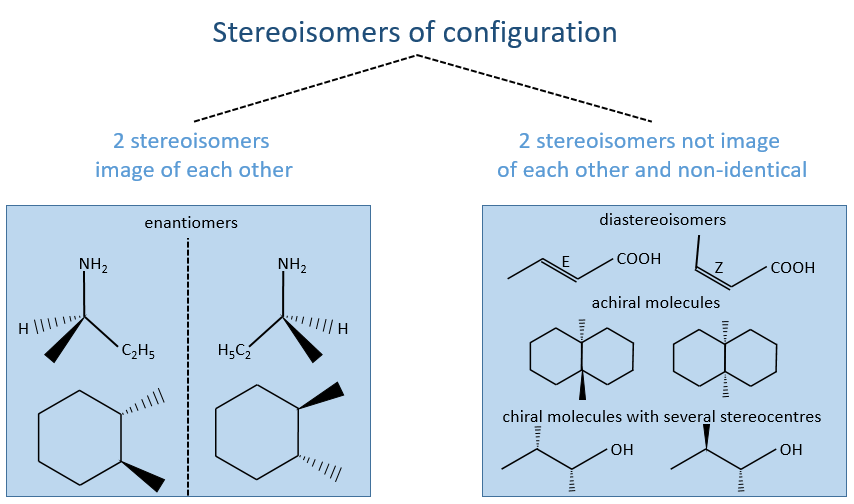
Amongst the stereoisomers of configuration, we distinguish the enantiomers that are chiral and the diastereoisomers that are not chiral and not identical.
Enantiomery
The distances between the atoms are identical in enantiomers. As a result, the chemical and physical properties of enantiomers are identical as long as there is no external chiral influence. A particular property of chiral molecules is their optical activity or rotatory power [α]. A chiral substance crossed by a polarised light induces a rotation of the plane of polarisation of the light in one direction.
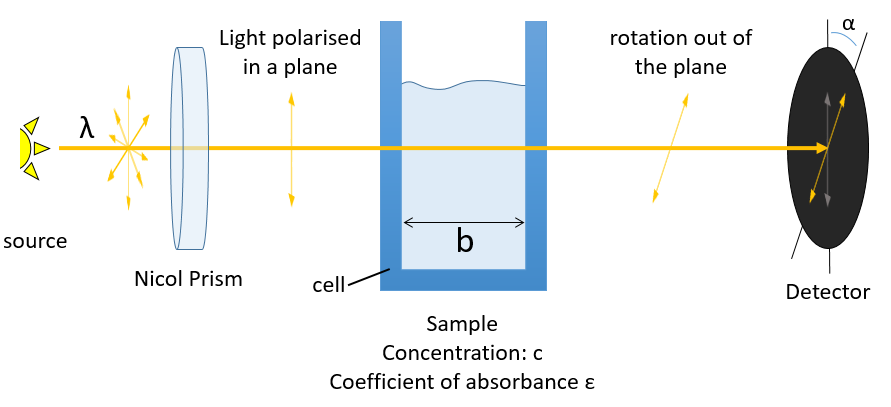
The direction in which the light is deflected depends on the enantiomer. For instance the 1,1-aminophenylethane has two enantiomers the optical activity of which is [α]D=39 and [α]D=-39 if illuminated by sodium at 20°C (the D indicates these experimental conditions). The specific activity of one enantiomer is given by
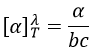
where λ is the wavelenght of the polarised light, T the temperature, α the observed angle of rotation, b the length of the cell containing the substance and c the concentration of the solution. The cell in itself has to have no optical activity.
If the value is negative, the compound is levogyre (noted l or (-)) and if the value is positive, the compound is dextrogyre (d or (+)). In the case of the 1,1-aminophenylethane, the R enantiomer is dextrogyre with an optical activity of [α]D=+39 and the S enantiomer is levogyre [α]D=-39). Unfortunatly, there is no correlation between the fact that the enantiomer is R/S and levogyre/dextrogyre.
If the solution is composed of a 50:50 racemic melange of two enantiomers, the light is not deviated and [α]=0. In fact the light is deviated at the miscroscopic scale but in average there is no deviation at the detector because the rotatory powers of the enantiomers counterbalance each other. We define the optical purity (also called the enantiomeric excess ee) as
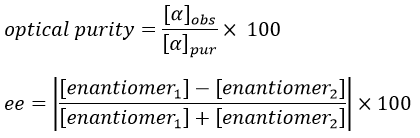
Stereogenic centres
1) centre of chirality
An asymmetric carbon (or any other tetravalent atom) on which there are 4 different substituents is a centre of chirality. The absolute configuration is the real disposition of the atoms around the atom of carbon. The carbon is S or R, what can be determined with the rule of Cahn, Ingold and Prelog (CIP)(there is a second method that can be used if you prefer it).
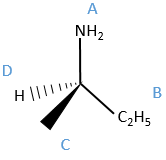
The first thing to do is to find the atomic number of the atoms bound to the assymetric carbon (consider zero for a lone pair). If two substituents have the same atomic number, we look at their substituents (starting with the highest atomic numbers), etc.
We can name them A, B, C and D starting by the highest atomic number. In the example above, the substituent A is NH2 because N has a higher mass than C or H, the other atoms directly bounds to the assymetric carbon. To determine B, there are two substituents bound via one carbon and one via one hydrogen. As the mass of C is larger than the one of H, we look at the substituents of the carbons, in the methyl group, there are 3H and in the ethyl there is two H and one C, so the ethyl group primes over the methyl group. We have thus A=NH2, B=C2H5, C=CH3 and D=H. The next step is to place the liaison between the carbon and the substituent of lowest atomic number in our axis of sight. The carbon is between us and the substituent D. The three other substituents are pointing towards us and are approximatively in the plane of the papersheet. We can draw the liaisons between them and the carbon.
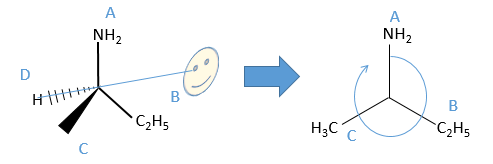
If A, D and C are in the horlogic order, the carbon is R. Otherwise it is S.
The second method uses the representation of Fisher on which the four liaisons are on the same plane. In reality it is not the case: to obtain this representation, imagine that you hold any two substituents with your thumb and your forefinger (for instance B and C).
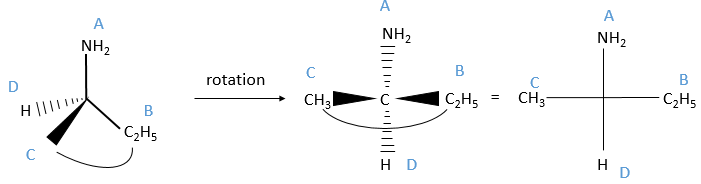
Then you rotate the molecule to form an horizontal plane with the assymetric carbon, pointing in your direction. The other two liaisons are vertical and pointing away from you. The goal now is to place the substituent of lowest priority at the top of the representation. To do that, we can swap the places of the substituents if the are direct neighbours. It is almost as simple as that: for each permutation made, the configuration also changes from R to S or vice versa. As a result, if we made two permutations, the configuration remains the same (R→S→R). We apply this at the end, but remember the number of permutations made.
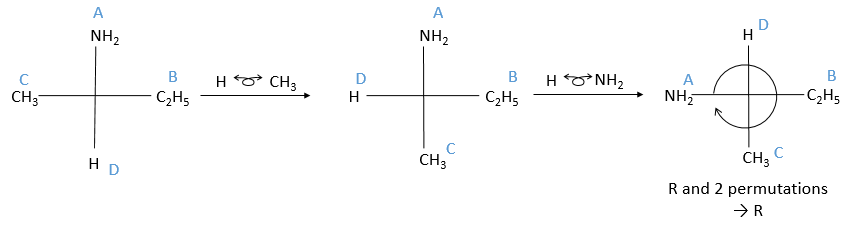
When the lowest priority subsitutent is at the top, we ignore it and we determine the “observed configuration” the same way than in the previous method: if they are in the horlogic order, then it is R, otherwise it is S. To obtain the real configuration from the observed configuration we just determined, we swap between R and S for each permutation made.
You may want to place the substituents in the horlogic order with permutation in all cases. You can do it if it helps you, but don’t forget that each permutation changes the configuration.
2) Axis of chirality
A similar method is followed. The difference is that we separate the substituents by their side of the axis ‘noted with and without prime). We will use the projection of Newton following the axis of chirality. The substituents at the front (the side we put in front does not matter) are noted a and b in function of their atomic number and the ones at the back a’ and b’. Next we check if aba’ is in the horlogic order (aR) or not (aS).
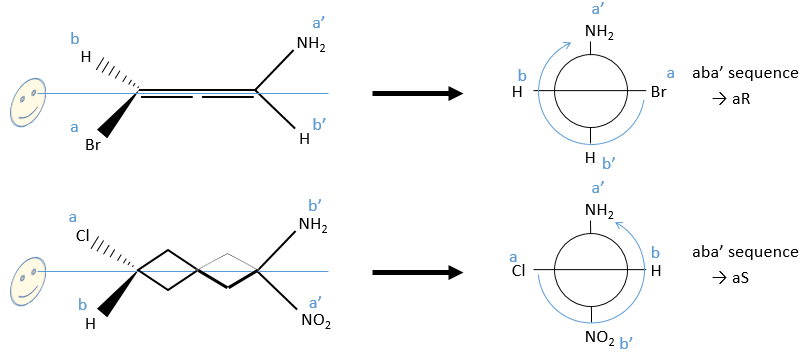
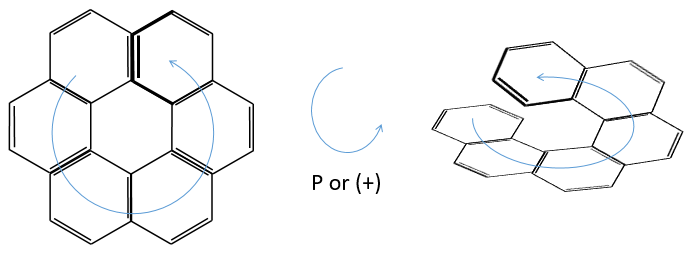
3) Helicoidal chirality
If the helix goes from the top to the bottom in the horlogic order, then it is noted P or (+). Otherwise it is M or (-).
Diastereoisomery
Two stereoisomers that are not enantiomers are diastereoisomers. It can be the case if there are several stereogenic centres in the molecule.
Two stereogenic centres
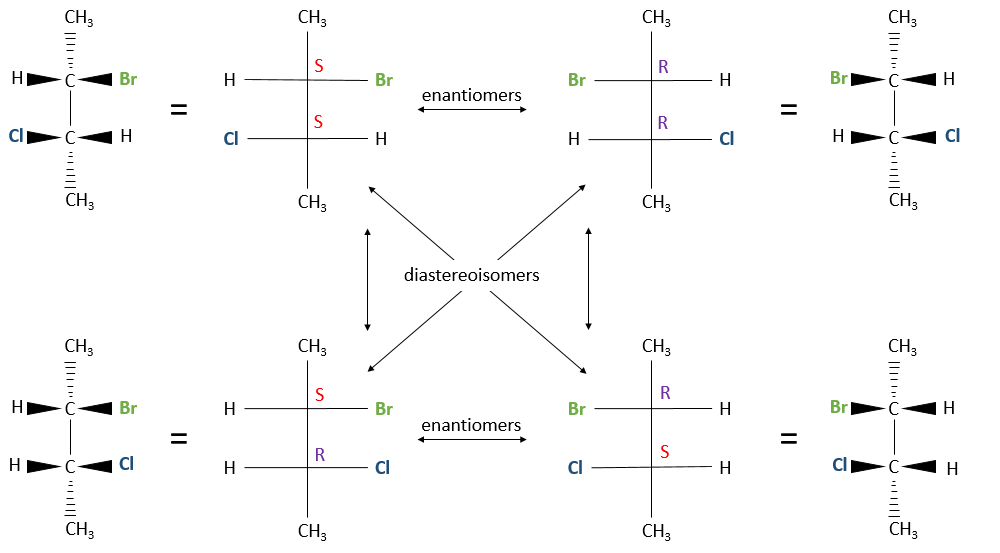
When the configurations are identical (RR or SS) are called like and when they are different (RS or SR), called unlike. Diastereoisomers haven’t the same chemical and physical properties because the interatomic distances are different. In the previous figure for instance, the distance between Br and Cl is different for the RS and the RR configurations. It is thus possible to separate them with usual techniques.
In the case of two asymmetric carbons, there is one enantiomer by configuration and two diastereoisomers. Yet the two diastereoisomers are sometimes one unique configuration that we call the meso form. For instance there is only one diastereoisomer for the enantiomers (2R,3R) and (2S,3S) of the 2,3-butanediol.
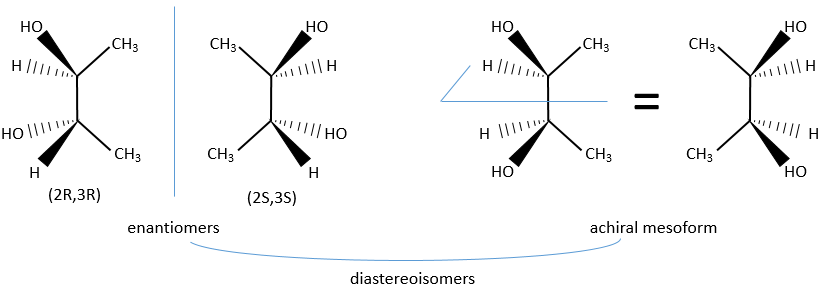
The reason here is a plane of symmetry between the carbon 2 and 3 of the butanediol.
Double liaisons
The presence of a double liaison entraves the rotation of the liaison. If there is at least one different substituent on each of the two carbons, there are then two stereoisomers that are called Z is the substituents of largest atomic number are in cis position, i.e. on the same side of the double liaison, and E if the substituents are in trans position, i.e. on the opposite side of the double liaison.
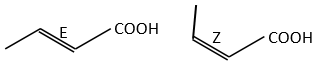
Prochirality
An achiral molecule is said to be prochiral if when we replace one of the substituent by a different one it becomes chiral. It implies that the two ligands that were apparently indiscernable are not equivalent.
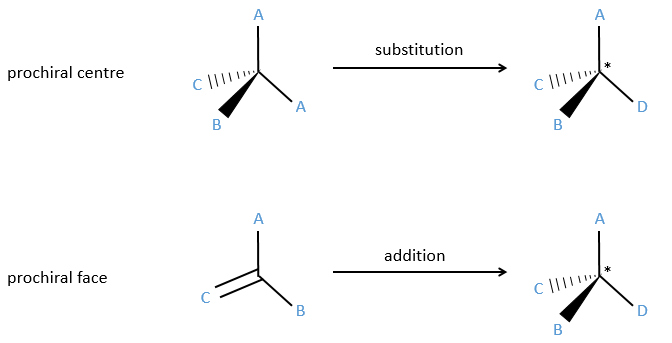
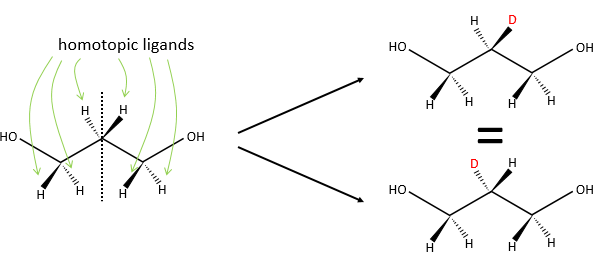
Homotopy and heterotopy
In a not-prochiral molecule, the substituents that are identicals and are related one each other by a rotation around an axis of symmetry of order n are said to be homotopic or equivalent. If we replace one of those equivalent substituent by a different one, the product is identical than if we replaced the other homotopic substituent.
In opposition, we talk about eniantiotopic ligands (or substituents) if there was a centre of prochirality. The two ligands are identical and are the result of a symmetry of each other but if we replace one of them by another substituent, then we don’t get the same molecule than if we replaced the other eniantiotopic ligand.
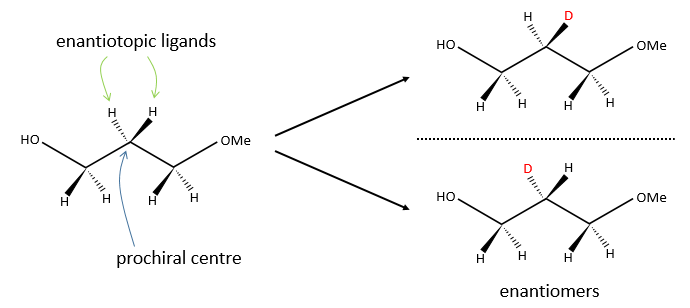
To name these molecules, we use the CIP method and we randomly give the priority to one of the eniantiotopic ligand. This ligand is pro-R if the CIP method gives R and pro-S if the CIP method gives S. The second eniantiotopic ligand has the opposite name.
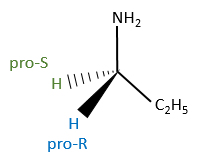
Two eniantiotopic ligands can’t be distinguished by usual physical and chemical methods, by RMN for instance. However, a chiral reactant like an enzyme can make the difference between the pro-R and pro-S ligands.
Eniantiotopic faces
In addition to eniantiotopic ligands, we also talk about eniantiotopic faces if a double liason is bound to a prochiral centre. The face of the plane on which the substituents are in the horlogic order is called face re and the other face is called face si.
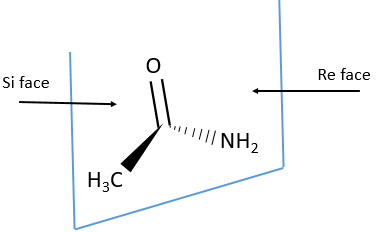
The probabilities to attack by the Re or by the Si face are equal if the nucleophilic species is achiral. In this case we obtain a 50/50 racemic melange of the enantiomers R and S. If the nucleophile is chiral, then the reactions on the Re and Si faces have different speeds. The reaction is diastereoselective.
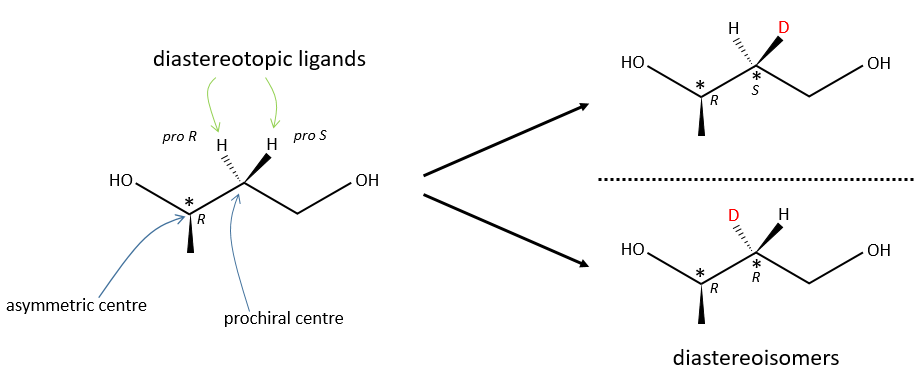
Diastereotopic ligands and faces
A prochiral centre can be near an asymmetric carbon. Two identical ligands of the prochiral centre are then not equivalent because they are not at the same distance from some groups of the asymmetric carbon. The replacement of one of the ligands or of the other leads to two diastereoisomers.
Achiral reactants can make the distinction between diastereotopic faces, one being usually more hindred than the other one. It leads to one product more formed than the other one. The products are diastereoisomers.
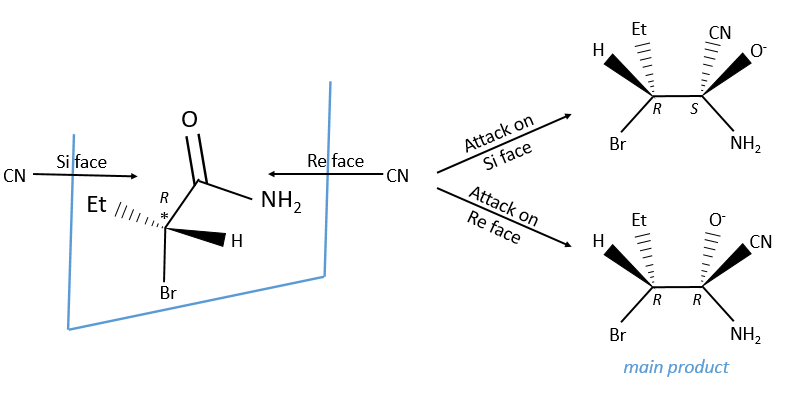
The less hindred side is usually favoured (model of Felkin).
Racemisation
It is the passage from one active optical body to the corresponding racemic melange. The racemisation can be due to time, heat or chemicals. In a general way, the cleavage of one liaison is involved and a carbon in the intermediate species possesses one sp2 carbon.
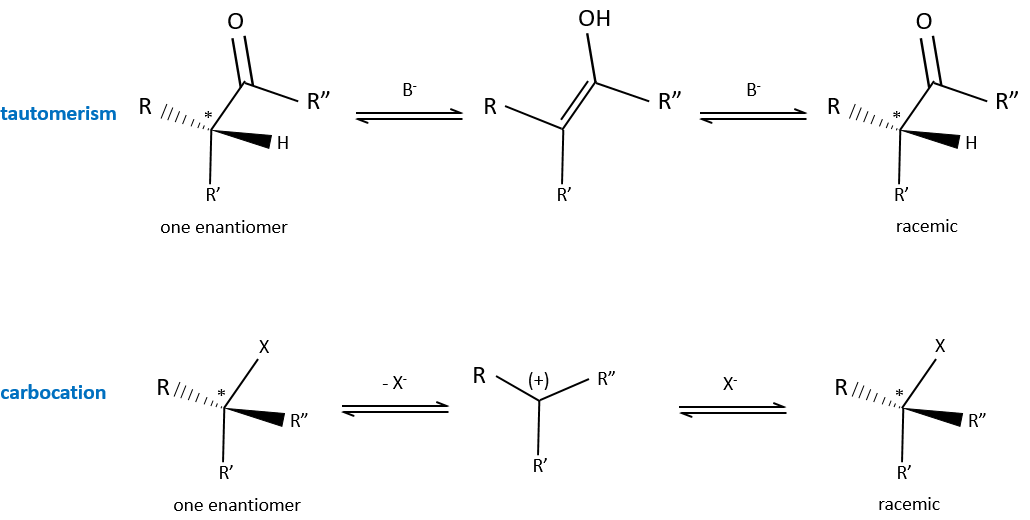
An example of racemisation is the enol-ketone tautomery. Starting from a solution made of one single enantiomer of a ketone, the other enantiomer will appear if there is a trace of a base in the solution because of the passage by the enol form. Under the sp2 form, the carbon is plane and there is thus no more S or R configuration. Both enantiomers can be formed from the enol.
Another example is the passage from one enantiomer to the other by the formation of a carbocation.
Epimerisation
Epimers are diastereoisomers with several stereogenic centres that differ only by the configuration of one of those centres. The epimerisation is the passage from one epimer to the other one.
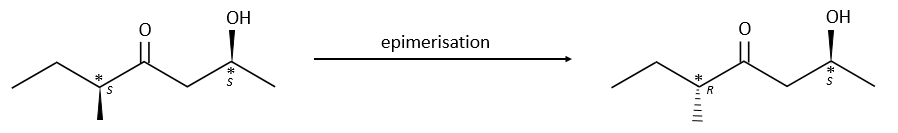
Chapter 2 : chemical kinetics – catalysis
Homogeneous catalysis
In some conditions, a same reaction A → B can lead to two different kinetics depending on the composition of the solution in which it is. One species C, apparently not intervening in the reaction as it is not consumed by it, increases the speed of reaction.
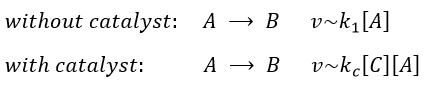
Both reactions occur simultaneously, giving a global reaction speed of
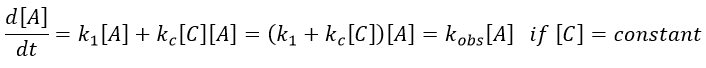
The ratio between the two paths is given by kc[C]/k1. The concentration of the catalyst can thus affect the contribution of the reaction paths. If the product of the reaction is the catalyst, then we say that the reaction is autocatalytic: the presence of the product enhances its own formation.
The use of a catalyst doesn’t change the difference of enthalpy and entropy between the products and the reactants but only the reaction path. The catalysed reactions involves several steps of smaller energy of activation than the normal reaction
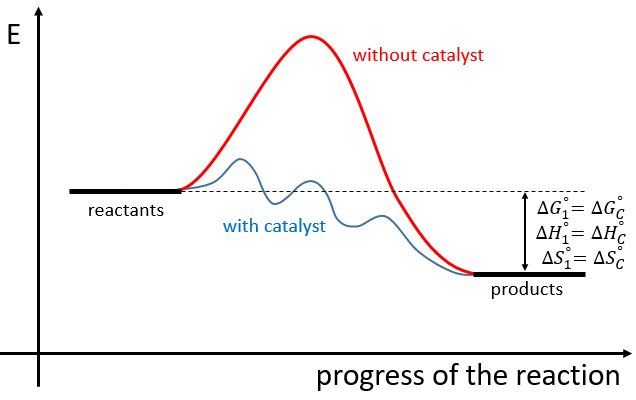
If the reaction is a reaction of equilibrium, the catalyst is catalyst of both sides of the reaction.
Electrophile catalysis – example of the hydrolysis of an ester
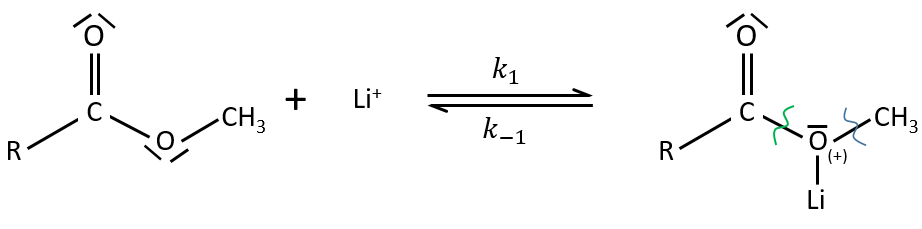
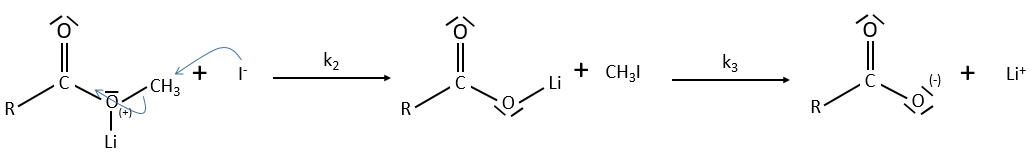
The steric hindrance can make the hydrolysis difficult. In this case we can use anhydric
LiI/pyridine to catalyse the reaction. Li+ is an electrophile that is complexed by the -O-. It acts as an acid of Lewis. The complexation eases the departure of R’ (-CH3 in the present case) which is normally a bad leaving group. K+ or Na+ can also be used.
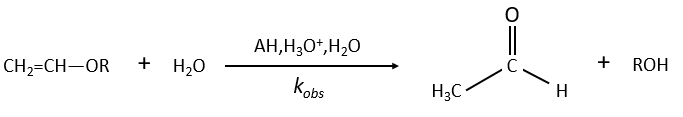
Specific acid catalysis
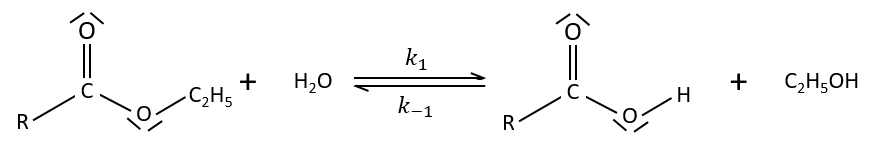
The formation of an acid A from an ester E is very slow in absence of catalyst. Proton can play the role of the catalyst in this reaction. The speed of the reaction will thus depend on the pH of the solution. The source of proton doesn’t matter but H3O+ is the catalyst.
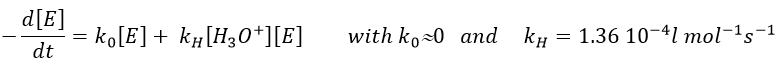
The reaction is reversible but we measure a pseudo order 1 if the acid is diluted: there is almost no product and the reverse reaction is thus negligible.
The concentration of the dissociated acid is calculated by the usual acid-base equation.
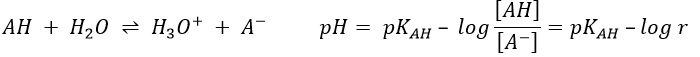
If [AH] varies at a constant pH, r is constant and thus kobs doesn’t depend on the concentration of AH. We are in the case of a specific acid catalysis.

The hydrolysis depends thus strongly on the acid-base equilibrium. The mechanism is
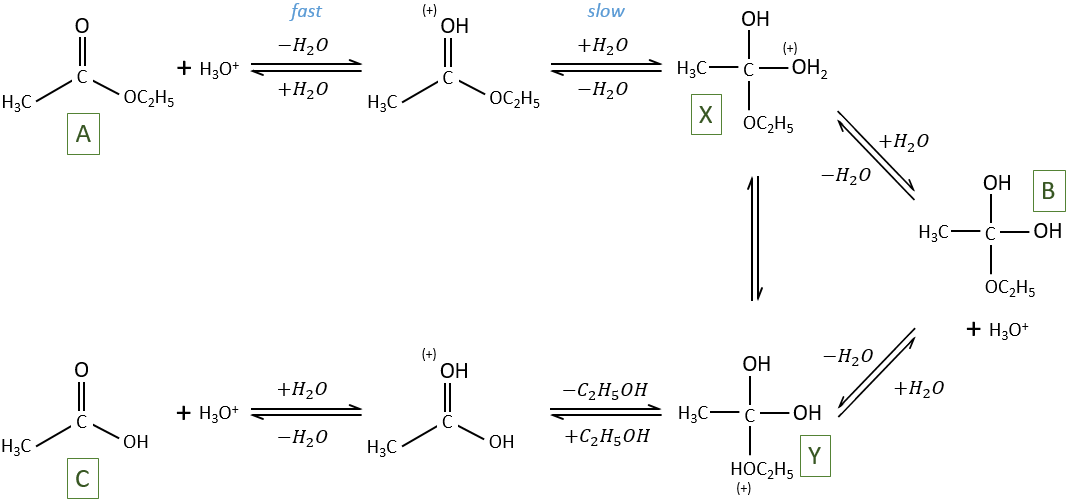
The protonation (first reaction) is fast and the nucleophilic attack by H2O (second reaction) is slow. X can turn into Y directly or first into B and then into Y.
General acid catalysis
An undissociated weak acid can also catalyse the reaction. If we have [H3O+] and [HA] constant, then
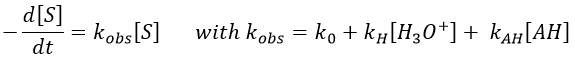
For a constant ratio [AH]/[A–], kobs varies with the concentration of [AH]. If there is a reaction involving AH, we have to recalculate [H3O+]. If there is no AH in solution, then we are back in the case of a specific acid catalysis.
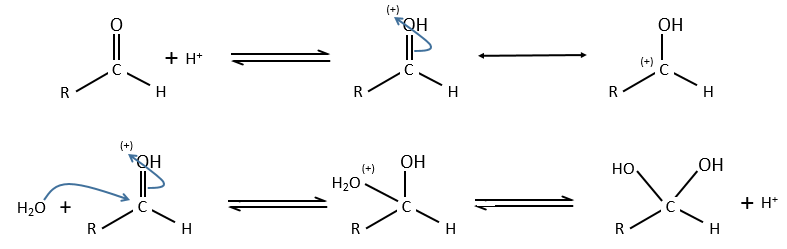
Example of the vinyl ether
The formation of an aldehyde and an alcohol from a vinyl ether (noted V) is catalysed by an acid. In a neutral or basic environment, the vinyl ether is stable (k0»0).
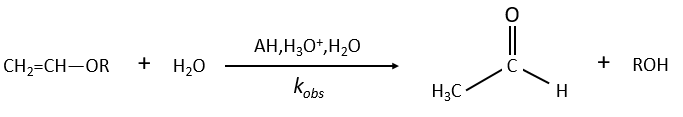

The protons are much more effective than the weak acid and it can be seen from the intensity of the constants of speed. For instance, if R=C2H5, kH= 2.08l mol-1s-1 and kacetic acid= 1.78 10-3 l mol-1s-1.
Comparison between specific and general acid catalysis: aldehyde into acetal reaction
In this case there is a difference of mechanism between the general and specific acid catalyses.
Specific:
Protons are directly involved in the first step, so the pH has an influence on the speed of reaction.
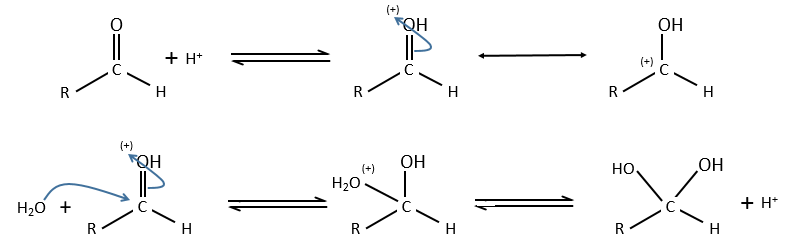
General:
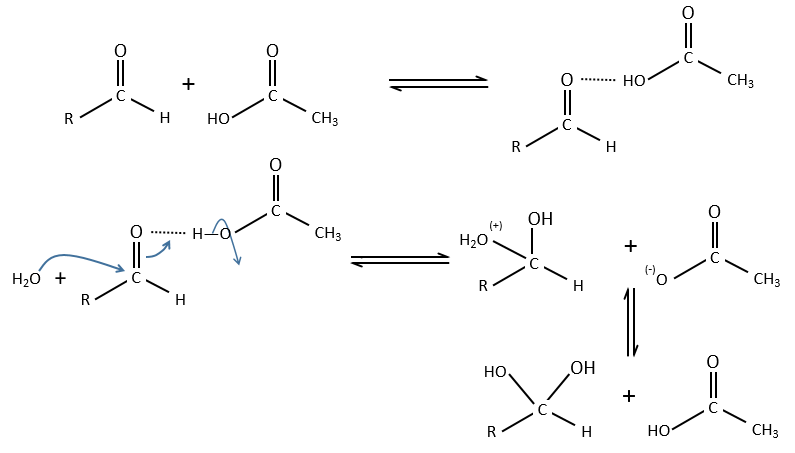
A weak acid forms an H bond to enhance the electrophile character of the carbon of the C=O and the transfer of proton is done simultaneously with the nucleophilic attack by the water. No proton are involved so the pH doesn’t change the dynamics. So to determine which catalysis is used, we change the concentration of the weak acid without touching the pH.
Nucleophilic catalysis
The catalyst interacts with an electrophile centre of the substrate. The process is slower and the intermediate species can be detected by spectroscopy or can be chemically trapped.
Example of the pyridine as catalyst
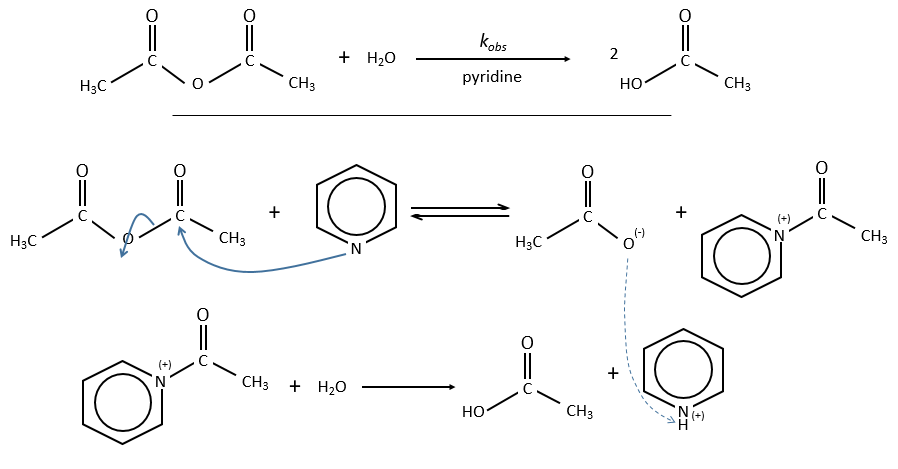
The acylpyridinium has a half-life time of 0.1s and can thus be detected.
Basic catalysis
Difference between specific and general basic catalyses
Specific:
General:
The NR3-H2O complex is more reactive than water because the lone pair of the oxygen is more nucleophilic.
OH– does not directly intervene in the general basic catalysis, as H3O+ does in the general acid catalysis. Yet the constant of speed depends on the pH because the general and specific catalysis are simultaneous. The general (basic or acid) catalyses are thus an important mechanism at pH’s close to 7, for instance in enzymatic reactions.
Enzymatic catalysis
It can be done several ways
- catalysis by proximity (entropic contribution)
- electrophile and nucleophile catalyses
- general basic catalysis
Catalysis by proximity
We compare here an intramolecular process with an intermolecular process.
Intermolecular
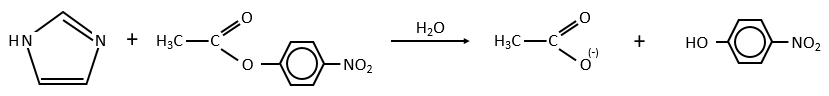
Imidazole is incorporated into many important biological molecules. One of them is the histidine that is present in many proteins and enzymes. The presented reaction is of the second order.
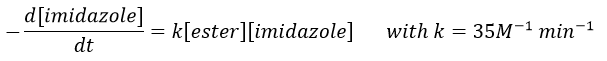
The p-nitrophenol is in equilibrium with its deprotonated form at neutral pH, form that can be detected via spectrometry of absorption.
Intramolecular
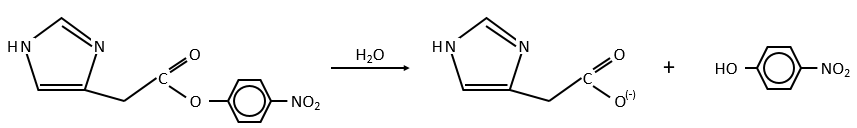
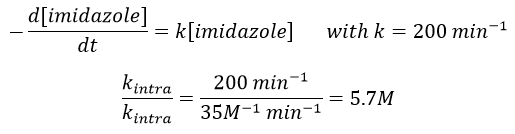
The fact that the catalyst was initially bound to the reactant is thus equivalent to the presence of 5.7M of the ester. The fact that the catalyst is already well positioned plays a role and there is also an entropic factor: from one molecule we produce two molecules and there is no need to bind the reactant to the catalyst anymore.
Nucleophilic catalysis
There are nucleophilic groups on enzymes. They can thus act as nucleophile catalysts (see previously).
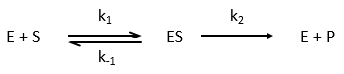
Equation of Michaelis-Menten
The enzymes are in small quantity in comparison to their substrates. The enzyme forms a complex with its substrate and then acts on it to form the product.
From the section on an equilibrium followed by an irreversible reaction, we know that
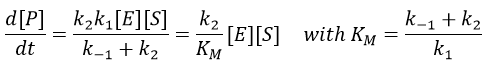
In this section, we have seen that there are two limit cases: the irreversible reaction is very fast or the equilibrium has time to be reached. In this case, the equilibrium has time to be established.
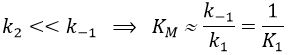
The quantity of enzyme is conserved over the time, in its lone form or complexed with the substrate.
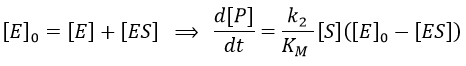
We used the hypothesis of stationarity on [ES].
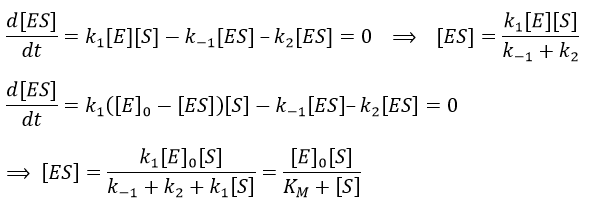
The formation of the product P is given by
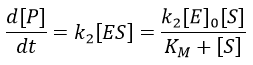
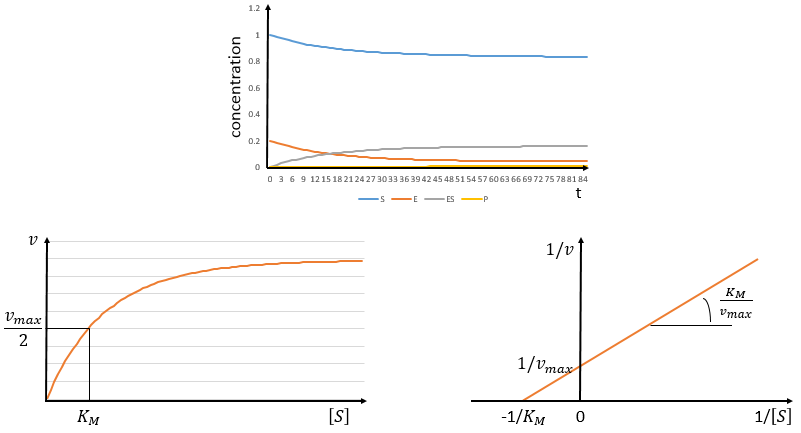
The speed of the reaction depends thus strongly on the concentration of the substrate up to a given point where [S]>>KM. The speed reaches then a plateau at v=k2[E]0. The maximum of the speed is thus reached when all the enzymes are active simultaneously ([ES]=[E]0).
![]()
The half of this speed is reached for a concentration [S]=KM.
By plotting 1/v vs 1/[S] (before the plateau), we find a straight line from which we can determine KM and k2.
If we consider one more step in the reaction, the formation of the product when it is still complexed with the enzyme, i.e. before the separation of the species, we will have a correction factor γ to add.
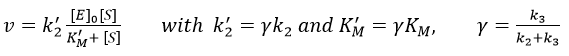
Case of the α-chymotrypsin
This enzyme is active in the digestive system where it hydrolyses polypeptides. It is active in this basic environment (pH between 8 and 9) and breaks the amide bonds between the peptides.
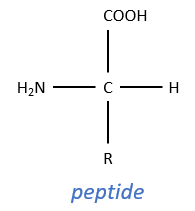
For the experiments, we substitute the polypeptides by an ester, the p-nitrophenylacetate.
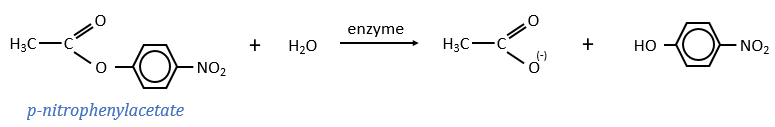
The p-nitrophenol is detected due to its yellow colour (405nm) and the acetate is not detected. The observations of the experiment are particular: the enzyme acts very fast at the beginning of the experiment and then the speed reaches a constant level. It is thus the opposite of what we have seen just above. If we look at the small times, the production of phenol is directly proportional to the quantity of enzyme. It seems that it is in this step of the reaction that we have vmax=k2[E]0. There should be a step after the release of the phenol that inhibits the enzyme. This step is the release of the acetate which is much slower than the release of the phenol.
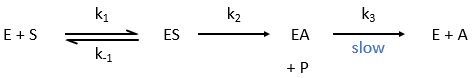
where P=p-nitrophenol, A=phenylacetate and k3<<k2.
As a result, when the phenol is released, the enzyme is not yet available to cleave another substrate. It has first to release the acetate, which is much slower. At the beginning of the reaction, we observe an important and quick increase of [phenol] that is quickly released by the enzyme. Before this moment, all the enzymes can do their job, what explains the large speed of reaction. After, one fraction of the enzymes is still occupied by one fragment of the substrate and cannot react until the acetate is released. The speed of the reaction is thus limited by the third step of the reaction.
We can assume that there is a nucleophilic site on the enzyme where the substrate binds at the level of the carbonyl.
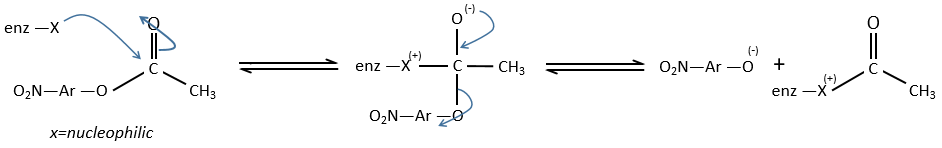
We can show that the intermediate acylenzyme is correct with the use of a more hindered substrate.
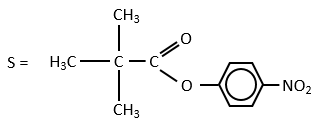
The steric hindrance and the inductive character of the methyl’s slow down the reaction, and thus they increase the life time of the acyled intermediate (~200 minutes). We can isolate the intermediate as crystals and analyse them.
We can use a better nucleophile than water to eliminate the acetate faster.
Chapter 1: chemical kinetics – relaxation to the equilibrium
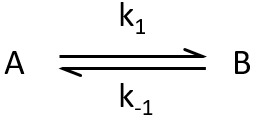
Reaction of order 1
Considering an equilibrium between two species A and B
We can study the return to the equilibrium (or relaxation) after a sudden modification of the conditions such as a jump of temperature or of pressure, or the modification of an electric field. The kinetics of the relaxation towards the equilibrium adapts to the new conditions.
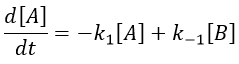
The concentrations can be noted as perturbations Δ[I] of the concentrations at equilibrium [I]e.
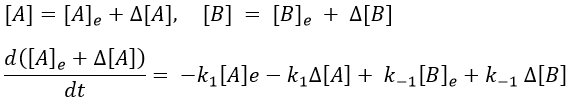
[A]e and [B]e don’t vary with the time. And at the equilibrium
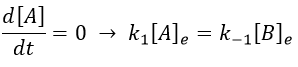
As the stoichiometric constants of the species are equal, we can write that
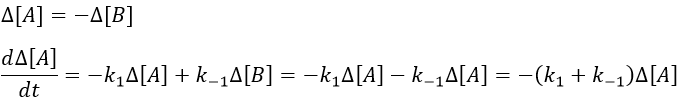
This equation can be solved.
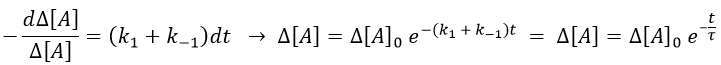
The relaxation is thus a decreasing exponential. The sign of the difference of concentration doesn’t matter: the equilibrium can be perturbed towards A or towards B, the relaxation will act the same way.
Reaction of order 2
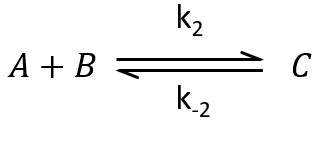
The methodology to determine the relaxation of this equilibrium is the same as for an order 1 reaction: we determine the speed of the reaction and insert Δ[X] in this expression. We use the properties that the concentrations are constant at the equilibrium and that only a small perturbation is applied to the system. This last assumption leads to a simplification of perturbations of order 2 or more (such as Δ[A]2 or ∆[A]∆[B]).
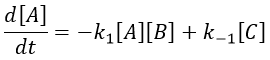
At the equilibrium, we have
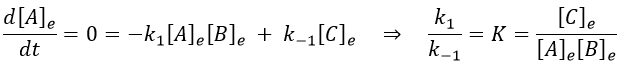
Inserting the perturbations in the expression of the speed, we obtain
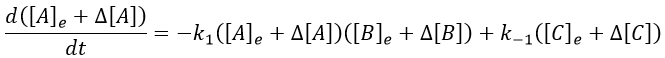
In the development of [A][B], we can ignore the ∆[A]∆[B] term because it is very small in comparison to the other terms. Knowing the stoichiometric coefficients, we know the relation between the perturbations in concentration.
![]()
It leads to the following expression.
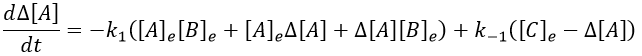
We can simplify the expression with the relation between the concentrations at the equilibrium.
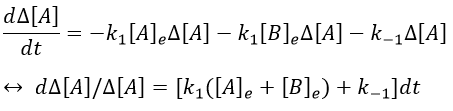
As a result,
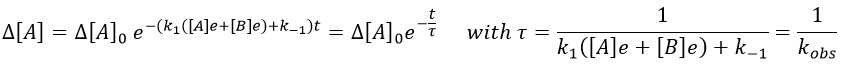
We can thus find the constants of speed of the reaction from the relaxation of the system that follows a perturbation. To do so, we repeat the experiment with several compositions of reactants. On the graph 1/τ vs [A]+[B], k1 and k-1 can be determined.
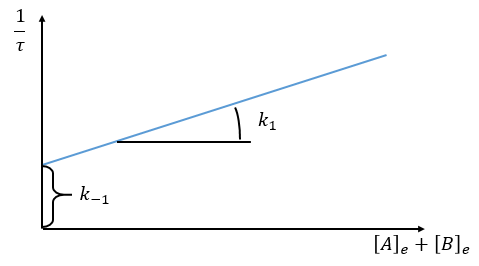
Optical measurement of the distance to the equilibrium
The relaxation is thus given by
![]()
If we want to follow the process optically, we can measure the absorbance α of the solution at a given wavelength.
![]()
It can be difficult to find a wavelength where only one species absorbs and not the others so we simply measure the absorbance of the solution at one wavelength even if it is a combination of the absorbance of several species.
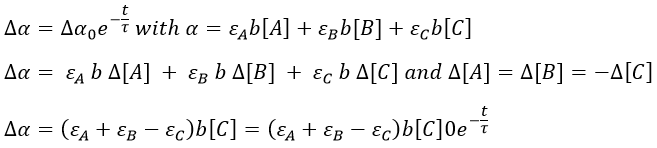
The precision will be the best if εA+εB-εC is large. We can check the intensity of the signal while at the equilibrium to find a wavelength for which the signal is strong.
Relaxation followed by conductivity
We can use this method of measurement if there is a perturbation of the ionic equilibrium of the solution. For instance in an acid-base reaction,
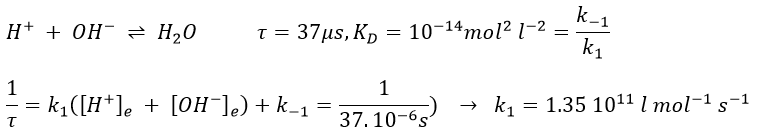
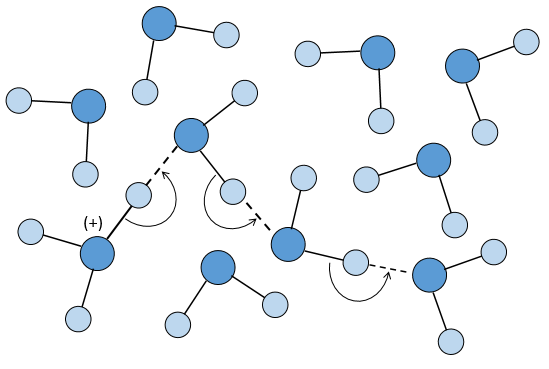
If the reaction has been driven by diffusion, we hould have found a relaxation period around 10-9-10-10 l mol-1s-1. The relaxation here is faster because the proton doesn’t properly diffuse. Instead, the proton-hopping phenomenon applies in aqueous solutions: each oxygen atom simultaneously passes and receives a single hydrogen atom that diffuses through the hydrogen bond network of water molecules.
Application: study of relaxation of the complex proflavin-DNA
The proflavin is a mutagen agent that can insert itself in the double helix of the DNA. It is an amphoteric species the heteroaromatic N of which is protonated at pH=7. The mechanism of interaction between the proflavin and the DNA is complex and not only driven by chemical processes.
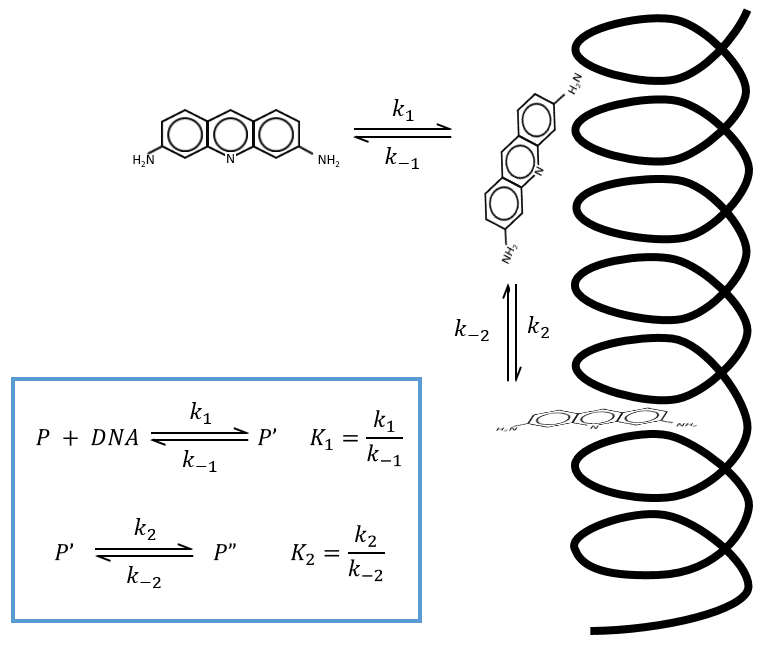
The supposed mechanism is that the proflavin diffuses near the DNA double helix in a first step. Once it is there, there is an interaction between the NH+ and the phosphates of the helix. In a last step the proflavin inserts itself in the DNA helix that had to open to let the mutagen agent enter. It may cause errors during the replication of the DNA by enzymes. We will note P the proflavin far from the helix of DNA, P’ along the helix and P” inside the helix.
If we impose a jump of temperature (~1micros) to the system when it is in equilibrium, we can measure the relaxation by absorbance. We observe that there are two relaxation times of different scales:
![]()
We can assume that there are two equilibriums involved in the process.
If the two reactions had been independent, then we would be able to use the expressions that we have find earlier in this chapter. However P’ is involved in the two equilibrium. Yet, the relaxation times are of different scales. If the first step is very fast in comparison to the second step, we can consider that the equilibrium K1 is reached. A quantity x of P and of DNA has been “consumed” in the first equilibrium (in the case of the DNA, consumed means that there is no place to bind with it on a given portion of the helix, one branch of DNA is capable to accept several proflavin molecules in its helix). A quantity z of P” is formed by the second reaction and a quantity y=z-x of P’ is consumed in total (y can be positive or negative).
![]()
As the first equilibrium is always reached over the relaxation period τ2 of the second reaction,
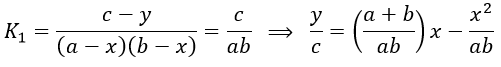
The x2 term can be neglected (small perturbation).
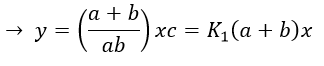
x=z-y so
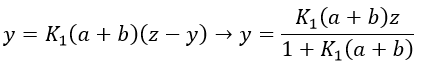
P” is involved only in the last step of the reaction which is slow.
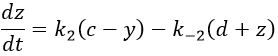
The value of y can be inserted in the equation to find that
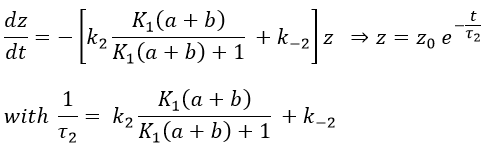
If the steps had been independent, we would have expected 1/τ2=k2+k-2 but there is a correction on the constant of speed k2 that involves P’ (which takes part in both reactions).
If K1(a+b)>>1, then the reactions can be considered as independent (1/τ2=k2+k-2)
If K1(a+b)<<1, then the relaxation time of the process is smaller than for independent reactions (τ2,coupled< τ2,indep)
If K1(a+b)<<<1, then the relaxation time is much larger than for the independent process (τ2»1/k-2).
We can play on the concentrations a and b to determine the constants of speed.
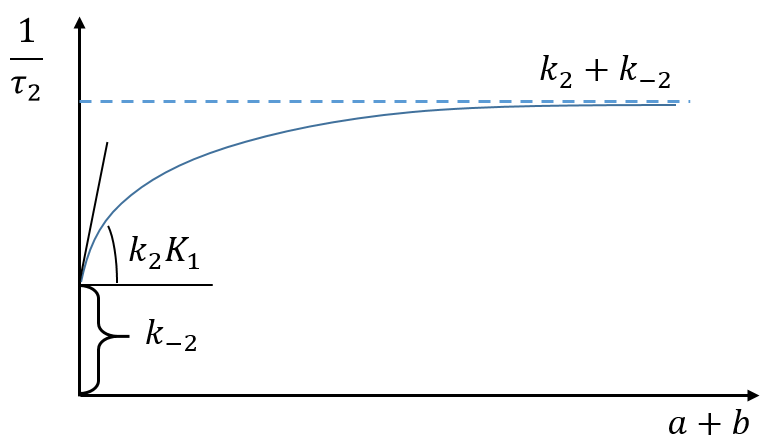
Experimentally, values have been determined for a pH=6.9, a temperature of 10°C and a concentration of Na+ of 0.2M:
k1=107 M-1s-1, a value corresponding to a diffusion driven process with a heavy molecule
k-1=3.5 103 s-1. This step is much slower than the diffusion because there is an electrostatic attraction between the helix of DNA and the proflavin. The dissociation is made without modification of the conformation of the DNA
k2=1.5 103 s-1 and k-2=1.1 103 s-1. We have a confirmation that the second step of the reaction is slower than the first step. It is longer to get inside the helix than to approach it. Note that it is slightly faster to move inside the helix than outside. The helix is disorganised and it is favourable from the entropic point of view.
Chain reactions
This section is useful for polymeric and catalysed reactions (for instance). In these reactions we discern three main steps: initiation, propagation and termination.
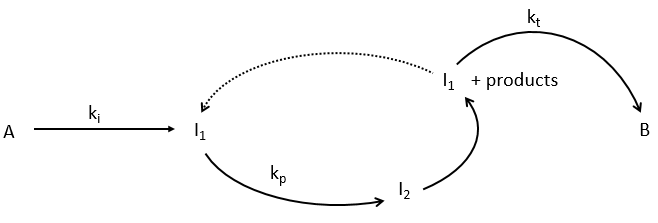
We will begin this section with the example of the formation of ortho-hydrogen from para-hydrogen (antiparallel nuclear spin and parallel nuclear spin). For this reaction (at 100°C), we find an order of 3/2.
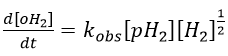
with H2 the total concentration of hydrogen (ortho and para). As soon as we see a concentration with a power of (2n+1)/2, we can assume that the mechanism is not elementary. The mechanism is thus more complex than a simple equilibrium. An additional species is required to proceed to the inversion of spin: a catalyst noted M.
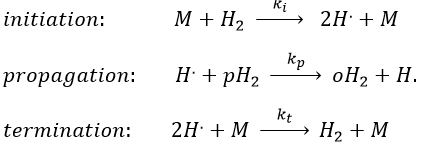
The propagation step can be repeated indefinitely as long as there is some pH2 in the system. Eventually, two radicals react together. The evolution of the concentration in radicals is given by
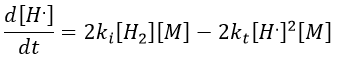
If we consider that [H.] is stationary, then we can find its value.
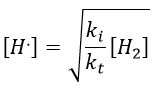
We can insert this value in the expression of the concentration of oH2.
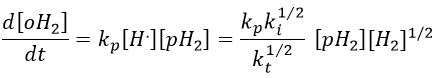
It corresponds to the experimental observation.
Example of HBr condensation
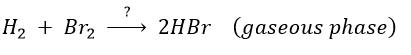
This reaction is not an elementary reaction and is in fact the result of a chain reaction. It is indeed hard to imagine that the two diatomic molecules that have not the same length of liaison, are perfectly aligned and vibrate in phase. The speed of the reaction can be determined experimentally:
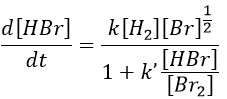
One can see that the product of the reaction is in the denominator of the equation, meaning that the speed of the reaction decreases with the concentration of the product. The explanation is that here should be an inhibition step in the process that consumes the product.
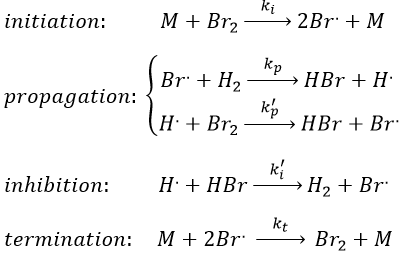
Note that the inhibition and termination steps are in fact the opposite reactions of the initiation and of the first reaction of propagation (kt=k-i and k’i=k-p). M is a catalyst on the surface of which the reaction occurs. Looking at the balance of the product, we have
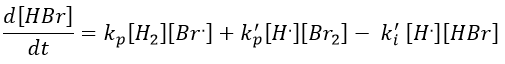
We will link this equation with the experimental expression of the speed by assuming the stationarity of the concentration of radicals H. and Br..
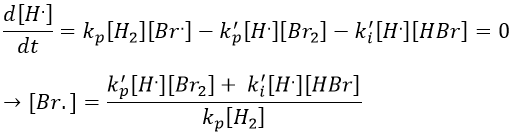
This expression still involves the concentration of a radical that is not measurable. We can insert this expression of [Br.] in the balance of the product.
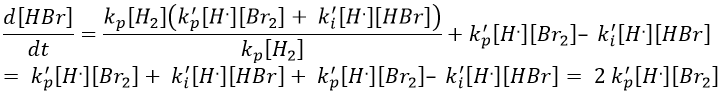
We do the same with the second radical.
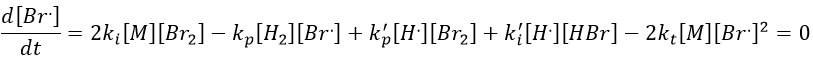
We can determine the concentration of Br. if we sum the variations of concentration of the radicals (still equal to zero):
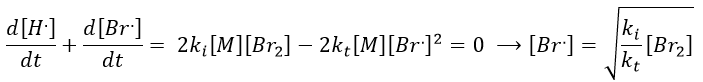
We can insert this expression in d[H.]/dt to obtain a measurable expression.
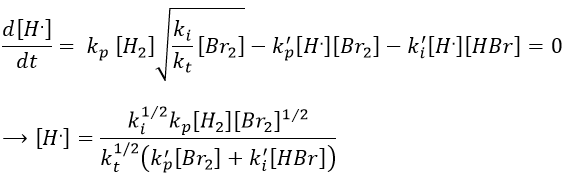
We can now express the concentrations of the radicals in the balance of the product HBr.
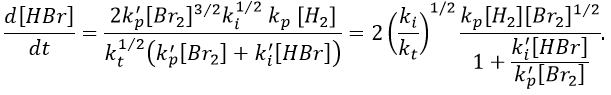
We can compare this expression to the experimental observation and conclude that
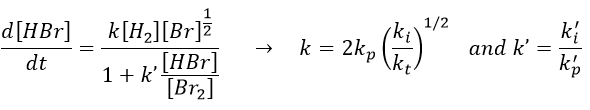
k’»0.1 is almost independent of the temperature (both reactions are exothermic and are of small energy). Note that some other reactions were possible and were not written. There is a second reaction of initiation and of inhibition but the energies of dissociation of H2 and HBr are way higher than the one of Br2.
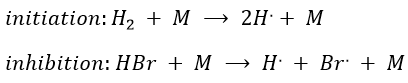
Some reactions of termination are also omitted for a different reason: the H. radical is extremely reactive and is active in the propagation step of the reaction. The concentration of this radical is thus very small.
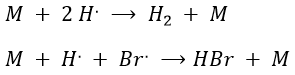
Note that those two reactions are the reverse reactions of the previous ones. Finally, there is also another method to initiate the reaction: instead of using a catalyst, we can use a laser (hν) but the termination step still requires M. As a result, the equations change and the kinetics are different.
This scheme of reaction is also valid for Cl2 and F2. In the case of I2, we observe an order 2 reaction kinetics. We have to heat up the system at 800K to obtain 95% of chain reaction.
Case of the stratospheric ozone
In the stratosphere at 20-40km of altitude, there is an equilibrium between the concentrations of O2 and O3. The rays coming from the outer space influence this equilibrium.
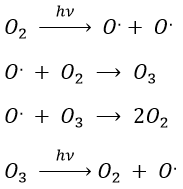
The ozone is important at this altitude because it filters the damaging UV rays from the sun. Before 1950, chlorofluorocarbons (CFC, for instance CFCl3, CF2Cl2) were commonly used as propellers in sprays and as coolers in fridges. No particular attention was done for their destruction and the CFC made a slow ascension towards the stratosphere. A hole in the ozone layer started to deepen at the poles (there because the hot ozone diffuses towards the cold areas). A problem is that the time it takes CFC’s to reach the stratosphere can be counted in years and when the phenomenon was detected (1971) a huge quantity of CFC’s were still on their way up.
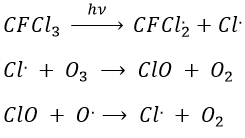
As a result, one portion of the ozone was consummed to form ClO and the equilibrium between O2 and O3 was displaced.